
文章摘要
分子氧活化以产生活性氧物种(ROS)是催化氧化偶联过程的核心,然而传统方法依赖外部能量输入,不利于可持续发展。本文首次证实,具有丰富自发生晶格畸变的多元素协同非贵金属高熵硫化物纳米晶(HESNCs),可在环境条件下自发活化氧气生成超氧自由基,以高催化活性与优异稳定性(>300天)驱动多种氧化偶联反应(S-N、C-S、C-N)。同时,该无外场驱动的HESNCs催化体系可高效合成药物分子(收率>90%),并可放大至摩尔级制备,展现出巨大的工业应用潜力。实验与计算分析表明,HESNCs中严重的本征晶格畸变可调控d带中心、增强泡利排斥作用,促使电子从有机给体(如胺类)转移至O2,生成给体自由基(Don·)。随后O2•−氧化底物形成偶联活性中间体(Sub·),与Don·结合得到氧化偶联产物,同时O2•−被还原为水。本工作建立了一种全新的“电子给体辅助-高熵催化剂(HECs)介导”的环境条件下O2活化范式,可在无外部能量输入下实现氧化偶联反应。
背景介绍
氧化偶联反应是有机合成中制备关键工业产品与药物的基础方法,通过外部氧化剂介导的电子转移实现亲核中心的连接。空气中的分子氧是氧化偶联反应的理想氧化剂,具有来源丰富、成本低廉、环境友好等无可比拟的优势。然而,环境条件下O2的实际应用受限于自旋禁阻反应路径。将O2活化产生活性氧物种(ROS,如单线态氧、超氧阴离子、羟基自由基)是克服O2固有缺陷、启动氧化偶联的有效策略。其中,O2•−因适宜的氧化还原电位(2.4 V vs. RHE)展现出优异选择性,显著降低过度氧化风险;且其半衰期长(51-422 s)、pH耐受范围广(pH=2-10),可在多样反应环境中保证稳定催化性能,成为可控氧化偶联反应的最优ROS。
结果与讨论
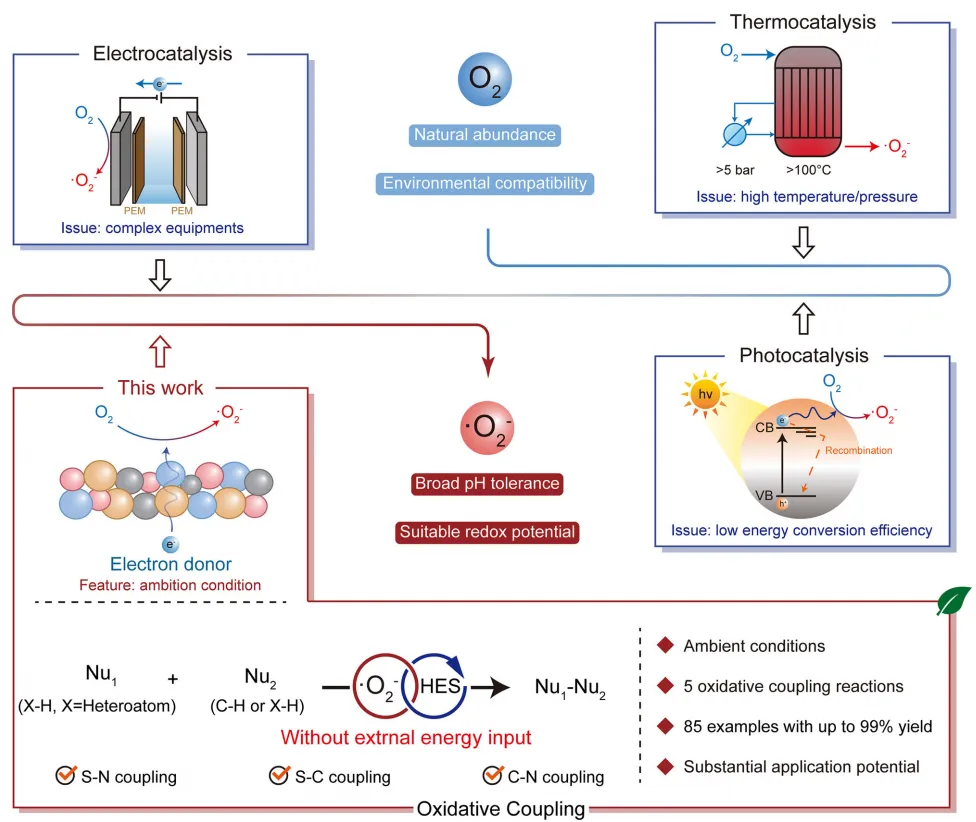
图1:构建策略。
直接将氧气活化生成超氧阴离子自由基,对于开发环境友好、成本低廉的工艺以实现常温常压氧化偶联反应极具研究价值。然而,氧气活化能垒较高,即便在催化剂存在条件下,超氧阴离子自由基的生成仍高度依赖外界能量输入(如光能、热能、电能),这导致反应操作复杂、效率低下且能耗偏高,极大制约了该工艺的工业化应用(图1)。从理论层面来看,通过催化方式直接将氧气活化为超氧阴离子自由基几乎难以实现:该过程需要发生大量电子转移,而电子转移通常会造成催化剂氧化,进而引发不可逆失活。因此,无需外界能量输入、直接活化氧气生成超氧阴离子自由基,以驱动常温常压氧化偶联反应,仍是一项巨大挑战。
芬顿反应可在常温常压下产生活性氧物种,该反应能够利用电子供体构建完整的循环路径,避免催化位点发生失活。在氧化偶联反应中,大量底物均具备作为高效电子供体的潜力。受此启发,本文报道了一项可持续催化领域的突破性研究:多元素协同非贵金属基高熵硫化物纳米晶体(HESNCs:FeCoCuZnCdS),其具有丰富的本征晶格畸变结构,可在无外界能量输入条件下实现氧气自发活化与底物氧化偶联。该高熵硫化物纳米晶体在常温常压下,对各类底物构建S-N、S-C、C-N键表现出优异的催化活性与稳定性(稳定性超300天),适配底物实例达85种以上;同时可实现摩尔级规模化合成,具备极佳的放大制备潜力,凸显其工业化转化应用的巨大前景(图1)。实验与理论计算分析表明,具有独特多元素协同效应的高熵硫化物纳米晶体,可介导电子从电子供体(反应底物)向分子氧转移,进而生成超氧阴离子自由基,并在常温常压下驱动后续氧化偶联反应进行。
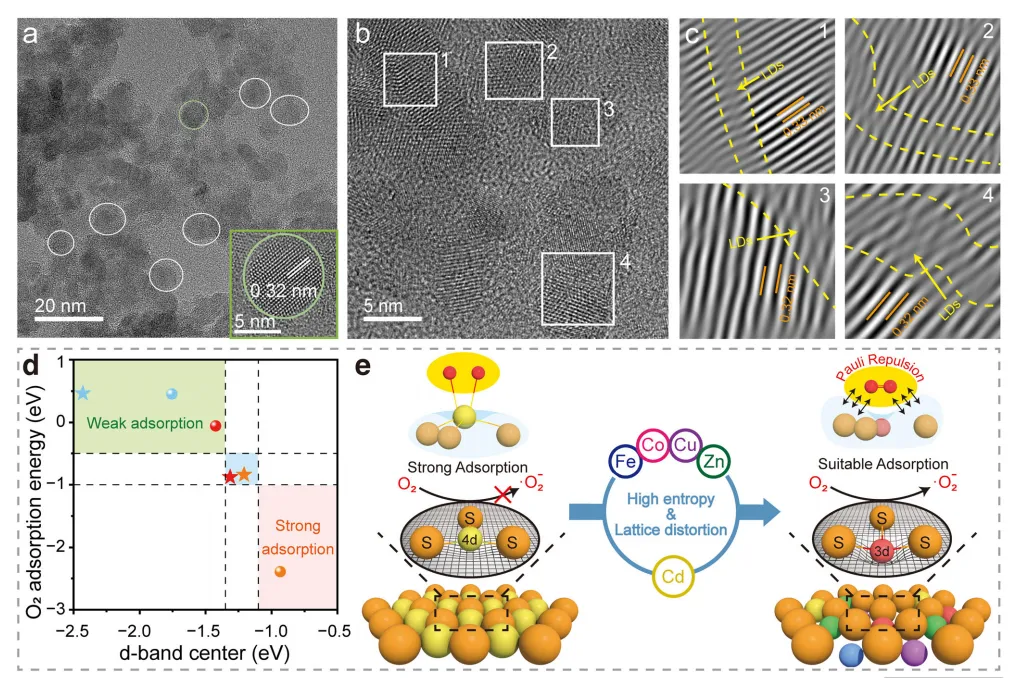
图2:铁钴铜锌镉硫高熵硫化物纳米晶体的结构特征与密度泛函理论计算。
粉末X射线衍射证实HESNCs为纯体心立方(BCC)结构。衍射峰明显偏移,与 Fe/Co/Cu/Zn选择性取代Cd导致的离子尺寸变化一致,证实形成具有显著压缩晶格畸变的单相固溶体。高晶格位错密度δ=2.40*1018 line m-2进一步验证了这一点。透射电子显微镜(TEM)图像显示,HESNCs为形貌规整的纳米晶,平均粒径6.5 nm(图2a)。高分辨TEM(HRTEM)测得HESNCs的晶面间距为0.32 nm,相较于纯CdS(0.34 nm)显著减小,证实存在严重晶格畸变。由HRTEM图像得到的逆快速傅里叶变换(IFFT)图谱(图2b、2c)为严重的本征晶格畸变提供了补充证据。
精准调控氧吸附能对高效生成O2•−至关重要:吸附过强会抑制超氧脱附,吸附过弱则无法活化O2。过渡金属催化剂与吸附质的相互作用强度取决于d带中心位置。因此,本文首先通过元素分态密度(PDOS)分析HESNCs的d带中心偏移,结果显示各元素d带中心发生显著偏移,表明相较于金属硫化物(MSs),HESNCs中发生了广泛的电子重分布。随后进一步研究d带中心变化与O2吸附行为的关联(图2d)。值得注意的是,HESNCs中Fe(吸附能=-0.84 eV)与Co(吸附能=-0.88 eV)具有接近理想的氧吸附能,这与其最优的d带中心位置直接相关。这种结构优化与HESNCs的富硫表面特征一致,该特征源于较小3d过渡金属掺入引发的晶格收缩(图2e)。这一发现与XRD衍射峰偏移及XPS中不饱和S(Sn2-)的存在相符。Sn2-丰富的孤对电子与O=O成键轨道Π产生显著泡利排斥作用,该排斥作用将O2吸附能从-1.30 eV 调控至-0.88 eV,进而促进活性氧脱附、避免催化剂失活。
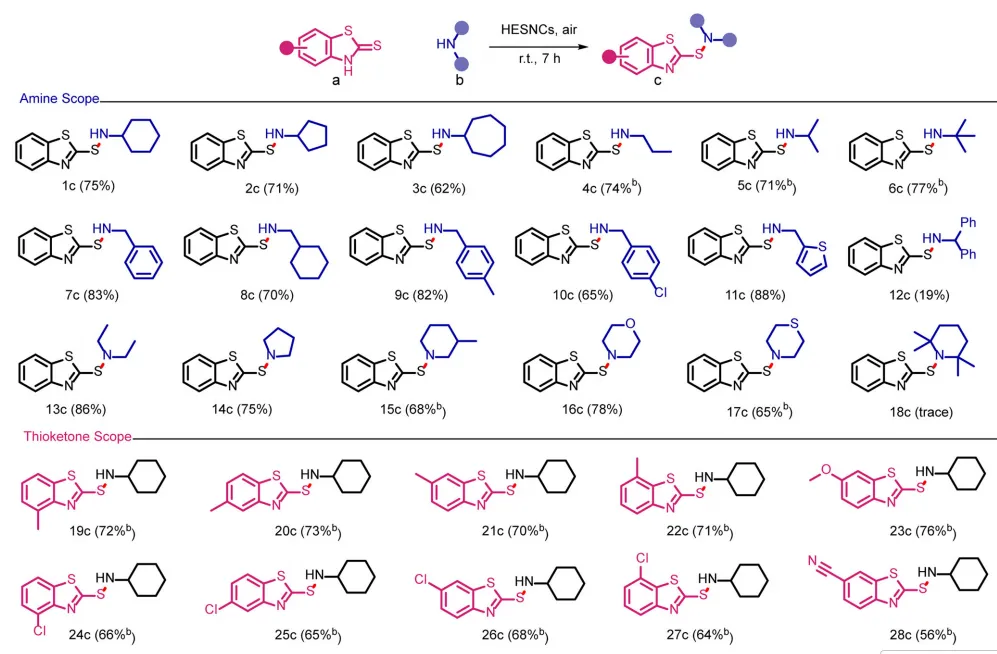
图3:高熵硫化物纳米晶体催化S-N氧化偶联反应的底物适用范围。
上述系统表征与理论计算证实,HESNCs具有严重的本征晶格畸变、最优的电子重分布与巧妙的富硫表面结构,具备在无外部能量输入下实现O2活化并驱动后续氧化偶联反应的巨大潜力。在众多氧化偶联反应中,胺与硫酮偶联制备次磺酰胺是合成高价值工业橡胶硫化促进剂(如N-环己基-2-苯并噻唑次磺酰胺(CBS)、N-叔丁基-2-苯并噻唑次磺酰胺(TBBS))最具价值的反应之一。前期研究证实O2•−可高效驱动S-N氧化偶联反应。因此,本文选取该反应为模型反应,评估HESNCs在无外部能量输入下的氧化偶联性能。以2-巯基苯并噻唑(1a)与环己胺(1b)为底物验证假设。反应条件研究证实,HESNCs可作为高效多相催化剂,在环境条件下生成O2•−并驱动氧化偶联反应。最优条件为:0.1 mmol(1.0当量)硫酮、0.5 mmol(5.0当量)胺、5 mg HESNCs催化剂,于2 mL乙腈中室温反应7 h。
在最优条件下,首先考察了不同伯/仲胺及位阻胺的底物适用范围(图3)。伯胺(链状、环状、苄胺取代,1c-12c)均以中高收率转化为目标次磺酰胺。而二苯甲胺(12c)收率仅19%,可能源于其较大空间位阻。以大位阻的2,2,6,6-四甲基哌啶(18b)为另一反应底物,进一步证实上述推测。其他低位阻仲烷基胺以良好至优异收率得到次磺酰胺13c-17c。此外,硫酮芳环上不同电子效应的取代基均具有良好耐受性(19c-28c),以良好至高收率得到对应S-N偶联产物。HESNCs催化S-N偶联的可放大性通过克级制备CBS(1c)得到验证,分离收率73%。HESNCs驱动的S-N偶联反应具有宽温度耐受范围,赋予其优异的气候与区域适应性。这些结果表明,HESNCs是首个可在无外部能量输入下实现可持续S-N偶联反应合成次磺酰胺的催化剂。
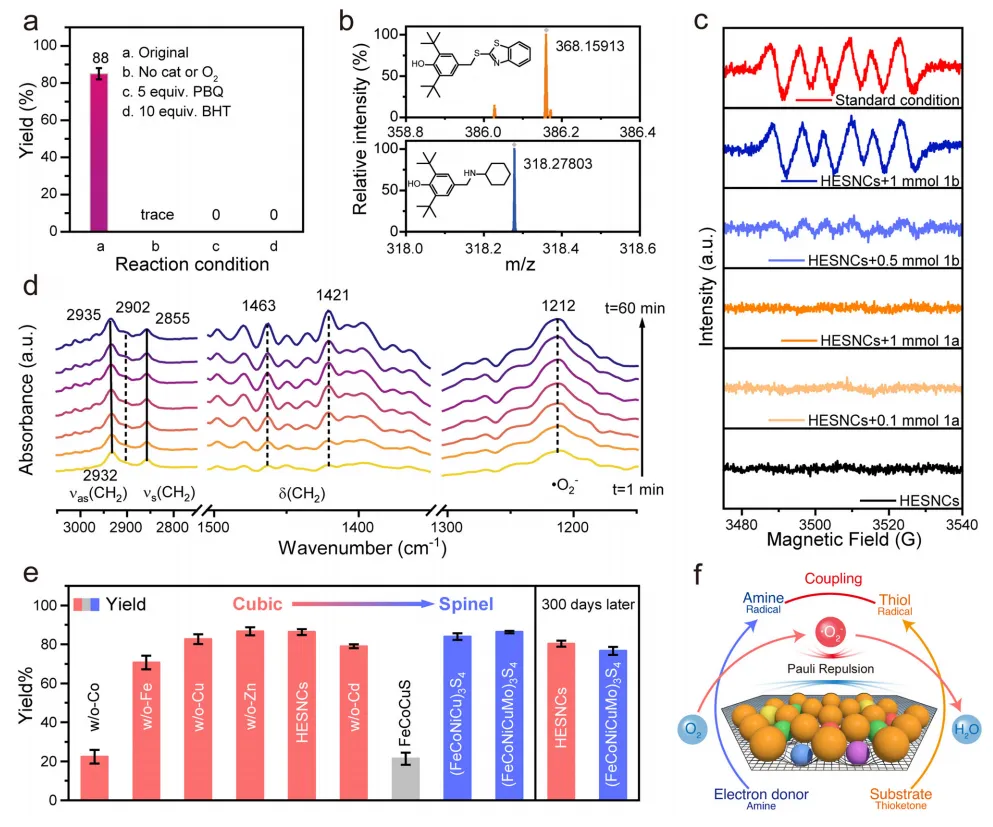
图4:反应机理研究。
为深入理解HESNCs驱动的S-N偶联反应机理,首先开展一系列对照实验(图4a)。结果表明,标准条件下排除O2或催化剂任一组分时,产物1c的生成量可忽略不计,确定二者在反应中的主导作用。随后进行自由基抑制实验:加入足量自由基猝灭剂2,6-二叔丁基-4-甲基苯酚(BHT)可完全抑制1c生成(图4a),证实反应的自由基本质。高分辨质谱(HRMS)观察到BHT与自由基的加合物,确认硫醇自由基与胺自由基为核心中间体(图4b)。此外,O2•−清除剂对苯醌(PBQ)可猝灭反应(图4a),表明O2•−是S-N偶联过程中的主要活性物种。为进一步探究O2•−生成过程,开展原位电子顺磁共振(EPR)实验(图4c)。首先,仅将HESNCs催化剂加入乙腈反应介质中,未检测到O2•−信号。随后分别向含HESNCs的介质中加入硫酮与胺。结果显示,硫酮存在时无法有效生成O2•−;而加入胺后,O2•−明显生成,且信号强度随胺浓度增加而增强,表明胺可在环境条件下诱导O2•−生成。这源于胺的给电子能力,其将电子转移至HESNCs,进而活化O2形成O2•−。更重要的是,标准条件下(5 mg HESNCs、0.1 mmol 硫酮、0.5 mmol 胺)检测到O2•−信号显著增强,表明硫酮可通过消耗胺自由基进一步加速O2•−生成。这些结果证实,本文建立了全新的“胺辅助-HESNCs介导”环境条件O2活化氧化偶联反应机理。
进一步通过原位/非原位傅里叶变换红外(FT-IR)光谱监测S-N氧化偶联反应过程。光谱显示,反应过程中CBS与O2•−逐渐累积(图4d),验证了“胺辅助-HESNCs介导”环境条件O2活化氧化偶联反应机理。此外,活性位点鉴定结果证实,HESNCs中的Co位点是主要活性位点,对促进O2活化与驱动氧化偶联过程至关重要。此外,中/高熵结构在环境条件下表现出高效的S-N氧化偶联性能(图4e)。值得注意的是,FeCoCuS的收率仅21%,表明催化性能与熵值直接相关。这一规律适用于不同晶相的Co基催化剂。结合上述实验与计算分析,本文提出无外场条件下电子给体辅助-HESNCs介导S-N偶联反应的完整机理:首先,胺(电子给体)向HESNCs表面发生单电子转移生成胺自由基,同时通过Co介导的电子转移将侧向吸附的O2活化为O2•−;随后,硫酮(底物)被O2•−氧化为硫醇自由基,自由基偶联生成目标产物,同时O2•−被还原为H2O(图4f)。
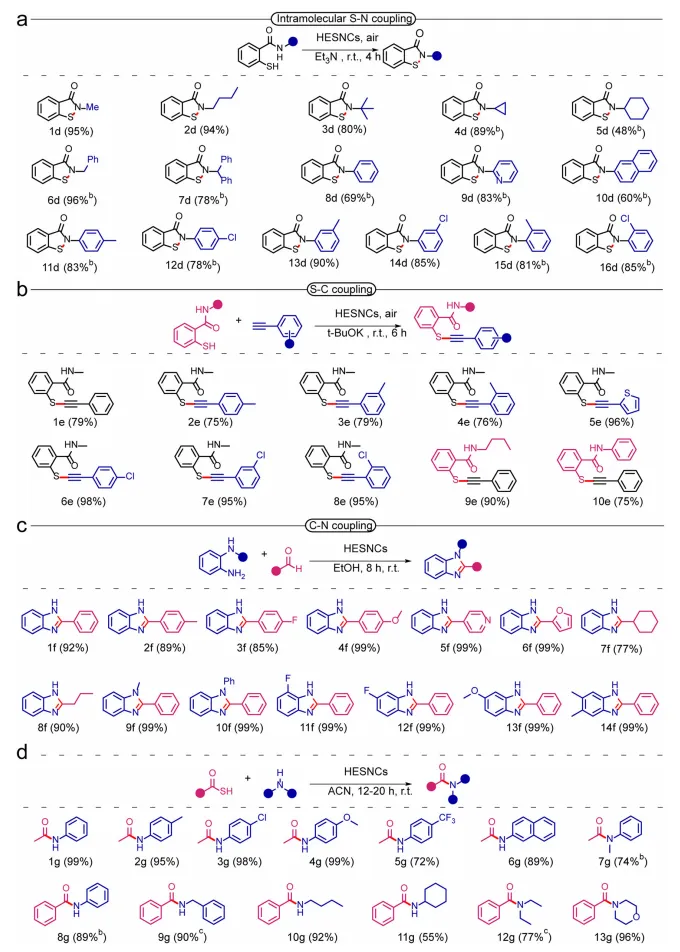
图5:高熵硫化物纳米晶体催化构建 S-N、C-S 及 C-N 键的产物收率。
为拓展“电子给体辅助-HESNCs介导”环境条件O2活化新机理的适用范围,将氧化偶联反应拓展至其他类型(图5)。苯并异噻唑酮是含S-N键的生物活性化合物,具有高效抗真菌、抗菌与抗精神病活性。本文结果表明,该体系可通过分子内S-N键形成,高效合成N-取代烷基或芳基1,2-苯并噻唑-3(2H)-酮化合物(1e-16e),收率60%–99%。鉴于C-S键在化学科学中的重要作用,硫醇官能化长期以来是药物化学与化学生物学的关键工具,在现代材料科学中也占据重要地位。因此,利用HESNCs通过2-巯基苯甲酰胺-苯乙炔偶联,实现了稳健的C-S键构建,底物取代基适用性广(1h-10h)。此外,C-N键是部分天然产物(如肽、蛋白质、壳聚糖)的基本骨架,其中酰胺与咪唑是有机化合物(如催化剂、药物、农用化学品、材料)中最重要的官能团。本文中,HESNCs催化剂成功介导醛与邻苯二胺之间的C-N键形成以合成苯并咪唑,对多种芳香醛(1f-7f)、脂肪醛(8f、9f)与邻苯二胺底物(10f-14f)具有广泛耐受性,生产高效、收率优异。此外,还实现了硫代乙酸与多种胺之间的酰胺合成,对芳基伯胺(1g-6g、8g)、芳基仲胺(7g)、烷基伯胺(9g-11g)、烷基仲胺(12g、13g)均具有优异底物通用性,且酰胺收率始终较高。HESNCs催化剂在分子内S-N、S-C、C-N偶联反应中表现出多功能性,同时保持广泛的官能团兼容性与底物适用范围。这些结果证实,“电子给体辅助-HESNCs介导”环境条件O2活化策略对氧化偶联反应具有普适性。
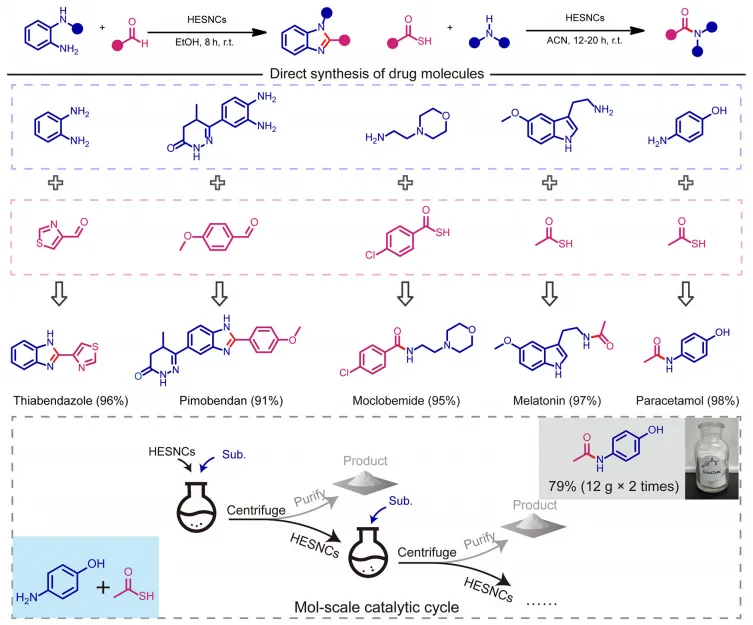
图6:药物分子直接合成及摩尔级放大实验。
随后,将该HESNCs催化体系拓展至药用功能生物分子合成,验证其应用潜力(图6)。多种生物分子,包括匹莫苯丹、噻苯达唑、吗氯贝胺、褪黑素与对乙酰氨基酚,均可通过环境条件下氧化偶联便捷制备,分离收率优异(>90%)。更重要的是,该催化体系通过两次连续0.1 mol 级循环,实现了对乙酰氨基酚的0.2 mol 级合成(24.1 g,收率79%)。这些结果表明,“电子给体辅助-HECs介导”环境条件O2活化新范式在工业级重要化学品合成中具有巨大应用潜力。
文章总结
本文开发了具有自发生晶格畸变与优化d带中心位置的非贵金属HESNCs,可在环境条件下自发活化O2并驱动一系列氧化偶联反应,无需外部能量输入。HESNCs在构建S-N、C-S、C-N键的反应中表现出优异催化活性(收率最高99%)、宽温度适应性(0℃以上收率无明显下降)、出色循环稳定性(多次循环后收率从88% 降至83%),并可放大合成药物(24.1 g,收率79%)。综上,本文提出一种通用的电子给体辅助-HECs介导O2活化新范式,可在环境条件下实现多样氧化偶联反应(S-N、C-S、C-N等)。该无外场催化体系展现出应用潜力。未来研究应重点关注:(1)反应放大与底物范围拓展;(2)开发可选择性生成其他ROS的HECs,以在环境条件下实现更多类型的氧化偶联反应。
文章信息
R.-L. Qi, Z. Guo, Y. Li, Y.-F. Ba, X. Xiong, T. Wei, M. Dan, Thiophene-based photocatalyst for exciplex-mediated singlet oxygen generation and H2O2 production.Angew. Chem. Int. Ed., 2026, e1664539.
文案:孙刚强

