太原理工大学蒙海兵/「国家杰青」张献明&北京邮电大学匡卓然&中科院上海高研院乔盼哲,最新Angew丨路易斯碱配位,高分散Pt高效光析氢!
合理优化Pt基助催化剂的配位结构,以同时实现增强的电荷分离/转移和降低的反应能垒,对光催化具有重要意义。2026年5月28日,太原理工大学蒙海兵、张献明、北京邮电大学匡卓然、中国科学院上海高等研究院乔盼哲在Angewandte Chemie International Edition期刊发表了题为"Lewis-Base Coordination Enables Highly Dispersed Pt Cocatalyst on Pyrene-Based MOFs for Enhanced Photocatalytic Hydrogen Evolution"的研究论文。Shuaiqi Guo、Haibing Meng为论文第一作者,蒙海兵、张献明、匡卓然、乔盼哲为论文共同第一作者。在此,作者通过接枝不同的Lewis碱基团,调控了Pt助催化剂在芘基金属有机框架(MOFs)上的配位。引入基团的软度直接决定了它们对Pt前驱体的亲和力,从而能够精确调控Pt与MOFs之间的电子金属-载体相互作用(EMSI),进而形成不同的Pt配位结构。这些结构特征通过建立内建电场加速电荷分离/转移,并通过氧化的Pt表面降低催化能垒,从而显著增强光催化活性。具体而言,-SH功能化的MOF(Pt/NUM)具有强EMSI,实现了Pt-O/S配位的原子级分散,在全光谱光照下使用抗坏血酸作为牺牲剂时,实现了5.68 mmol gcat⁻1 h⁻1(405.71 mmol gpt⁻1 h⁻1)的优异析氢速率,约为Pt/NU对照组的16倍,并超越了许多已报道的MOF基材料。值得注意的是,该催化剂在20小时连续运行中保持了卓越的稳定性。这项工作突出了Lewis碱配位在调控活性位点和优化EMSI方面的作用,为合理的催化剂设计和相关能源应用提供了新见解。氢能因其高能量密度和环境友好性,被认为是不可再生、CO2排放化石燃料的理想替代品。在这方面,已采用多种技术来实现析氢。其中,光催化水分解析氢因其低成本、高安全性和可行的催化效率而脱颖而出。
然而,其实际应用仍面临开发高效光催化剂的重大挑战。在光催化系统中,高度分散的金属物种(如单原子、团簇和纳米颗粒)通常被用作催化活性位点,它们可以影响光生载流子动力学以及反应能垒,从而决定整体光催化效率。
特别是,单原子金属位点因其最大的原子利用率而表现出极高的光催化活性,大大降低了相应贵金属的使用量,从而降低了相关成本。然而,这些活性位点由于高表面能而倾向于热力学聚集成大颗粒,导致催化活性不理想。因此,开发优异的基底来稳定和活化这些活性位点对于高效光催化析氢是可取且重要的。
据报道,增强电子金属-载体相互作用(EMSI)不仅可以改善活性位点和基底的电子和界面结构以及反应中间体的吸附/脱附,还可以促进电子传输,从而提高催化活性和选择性。因此,TiO2、MnO2、CeO2等许多基底已被改性以增强EMSI并锚定催化位点以实现高效析氢。
然而,这些半导体仍然存在表面锚定位点有限、光吸收差和严重的刚性体结构等问题,阻碍了性能的提升。因此,开发先进的基底来解决这些缺陷具有重要意义。
近年来,二维金属有机框架(2D MOFs)因其超薄的厚度、高长径比和大的比表面积而备受关注,这有利于金属位点的暴露和质量传输。此外,它们通过组装不同的金属节点和有机连接体以及后合成修饰,展示了多样化和可调控的结构。特别是,作为活性基底,它们可以固定单原子或纳米颗粒并确保其均匀分布。
由于这些结构优势,2D MOFs被广泛用于通过可控配位支持催化活性物种以实现先进的光催化应用。然而,目前关于2D MOFs的光催化研究主要集中在通过本征结构设计和杂化材料工程来解决低效的载流子分离和缓慢的电子-空穴转移动力学。相比之下,对它们上催化活性位点的分子级工程以及揭示增强光催化的潜在机制仍然探索不足。
关键的是,尽管配位诱导的EMSI在这些光催化系统中起着重要作用,但其基本机制仍然 poorly 阐明。具体来说,必须解决两个关键问题:具有不同电子性质的功能基团如何定量调控EMSI的强度?这种差异化的EMSI又如何分别和协同地优化电荷分离和反应能垒?
因此,在分子尺度上合理工程化2D MOFs上催化位点的配位环境以精确调控EMSI势在必行,这既是为了解决这些基本知识空白,也是为了建立明确的结构-活性关系。
在此,作者通过接枝不同的Lewis碱基团(-SH、-OH、-NH2)来调控Pt助催化剂在芘基MOFs上的配位,以促进光催化。研究表明,-SH配位最大化了Pt与MOFs之间的EMSI,有效触发了Pt的原子级分布,形成Pt-O/S配位,通过强化内建电场促进光生电荷分离/转移,并促进Pt的氧化以降低反应能垒。
因此,获得的Pt/NU-M在全光谱光照下表现出优异的析氢速率5.68 mmol gcat⁻1 h⁻1(405.71 mmol gpt⁻1 h⁻1),并在连续运行约20小时中保持高耐久性,超越了许多已报道的MOF基光催化剂。这项工作的核心创新在于将Lewis碱工程化概念化为调控EMSI的强大"分子开关"。
通过构建用-SH、-OH和-NH2基团合理修饰的对比系统,作者证明了强EMSI发挥着协同作用:它不仅稳定高负载的Pt单原子,还强化内建电场以优化电荷分离动力学,并提高Pt氧化态以降低反应能垒。此外,这项工作证明,通过靶向分子功能化进行配位工程为调控EMSI提供了可行途径,从而为设计高效MOF基光催化剂铺平了道路。
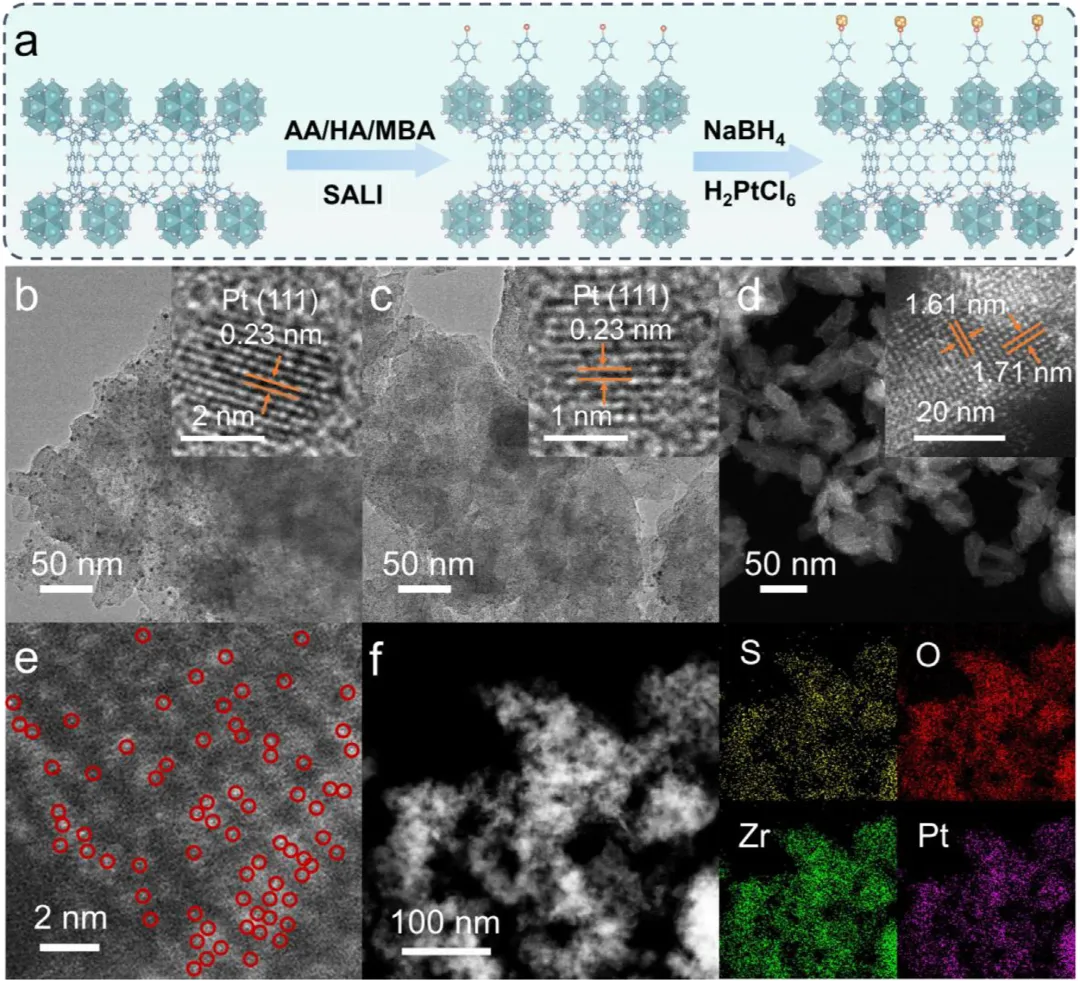
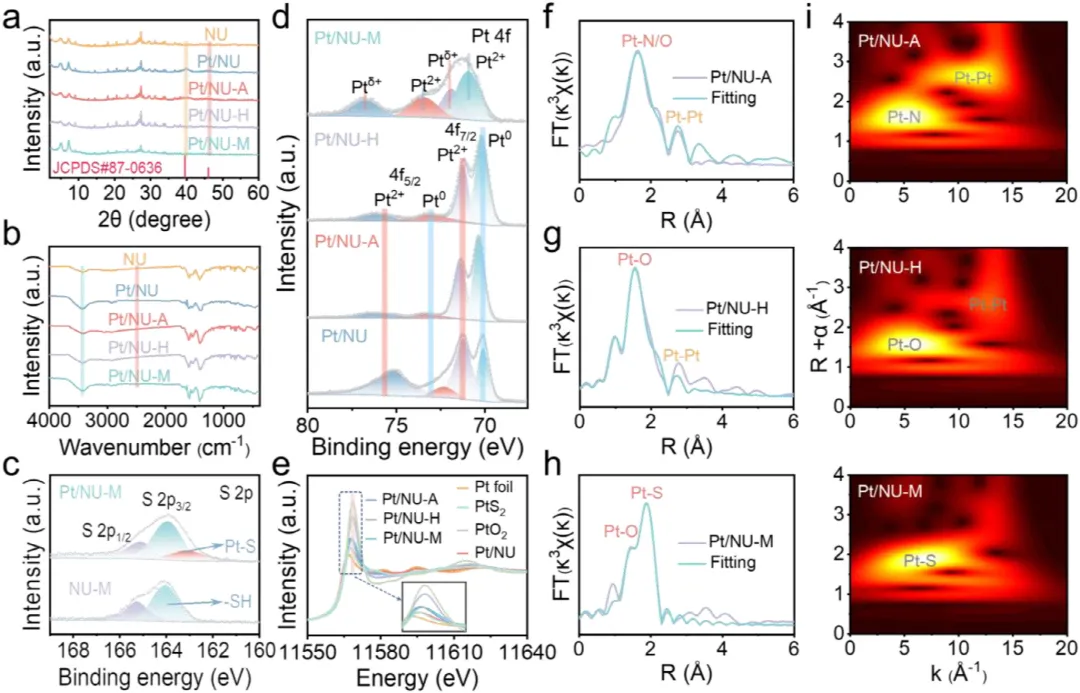
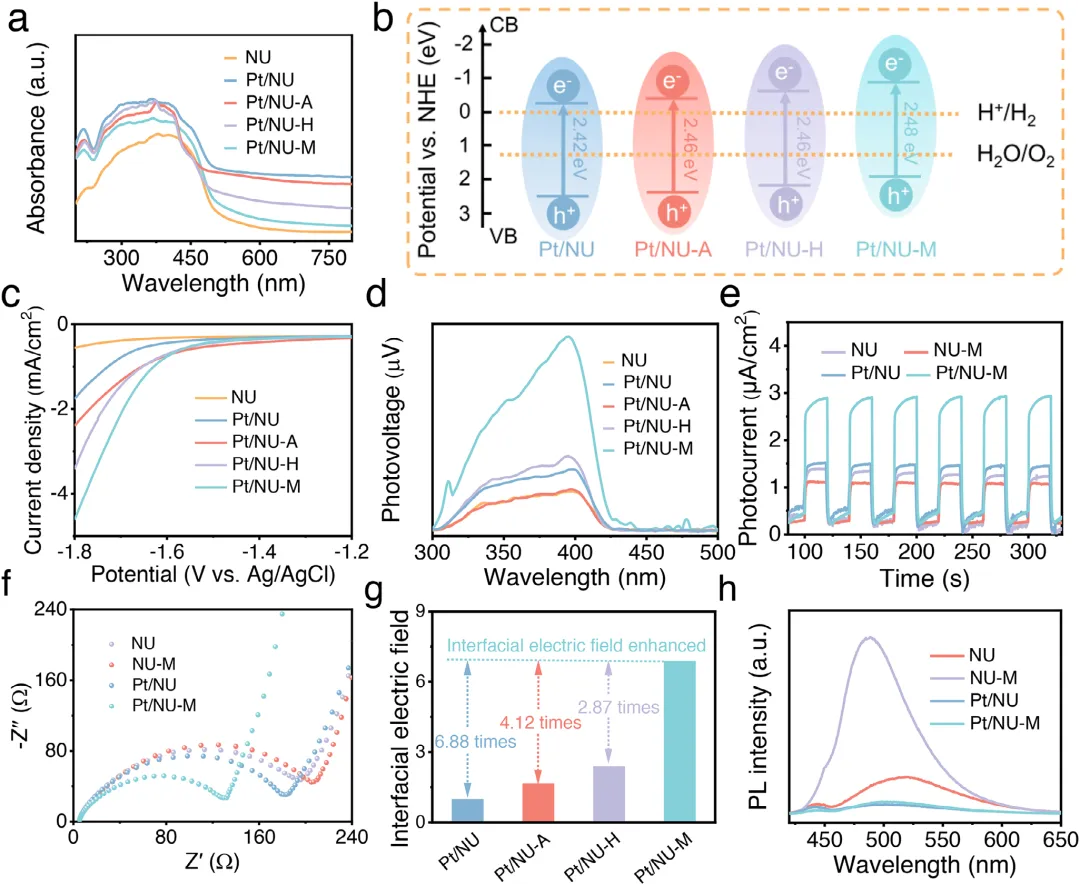
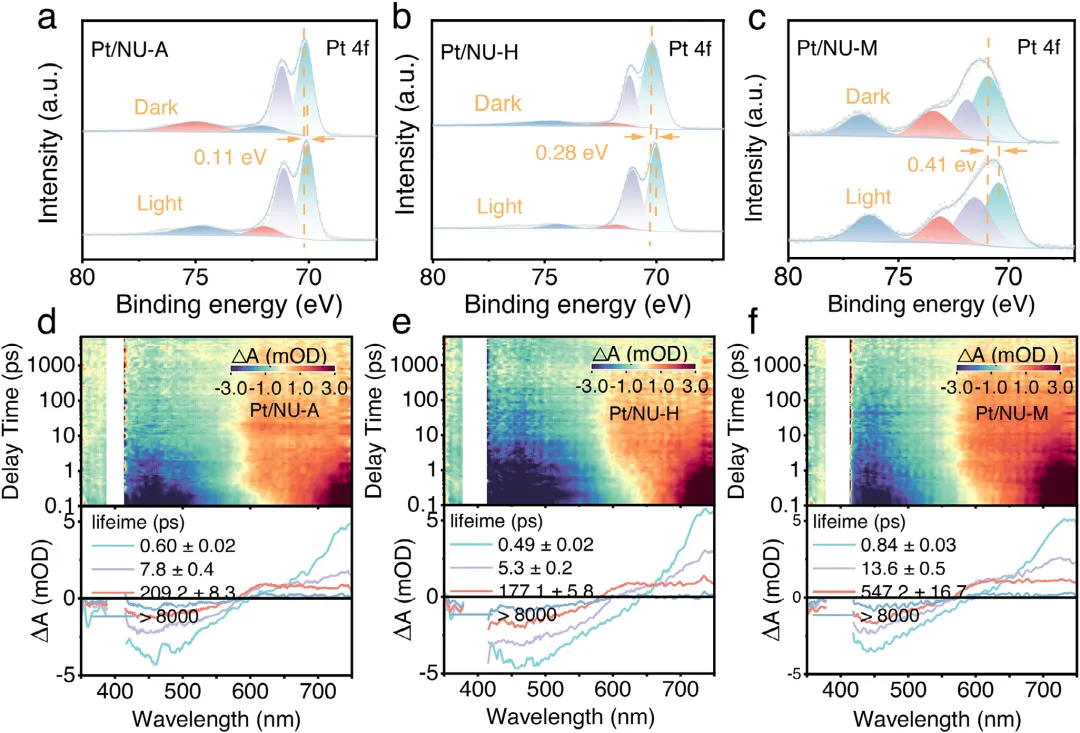
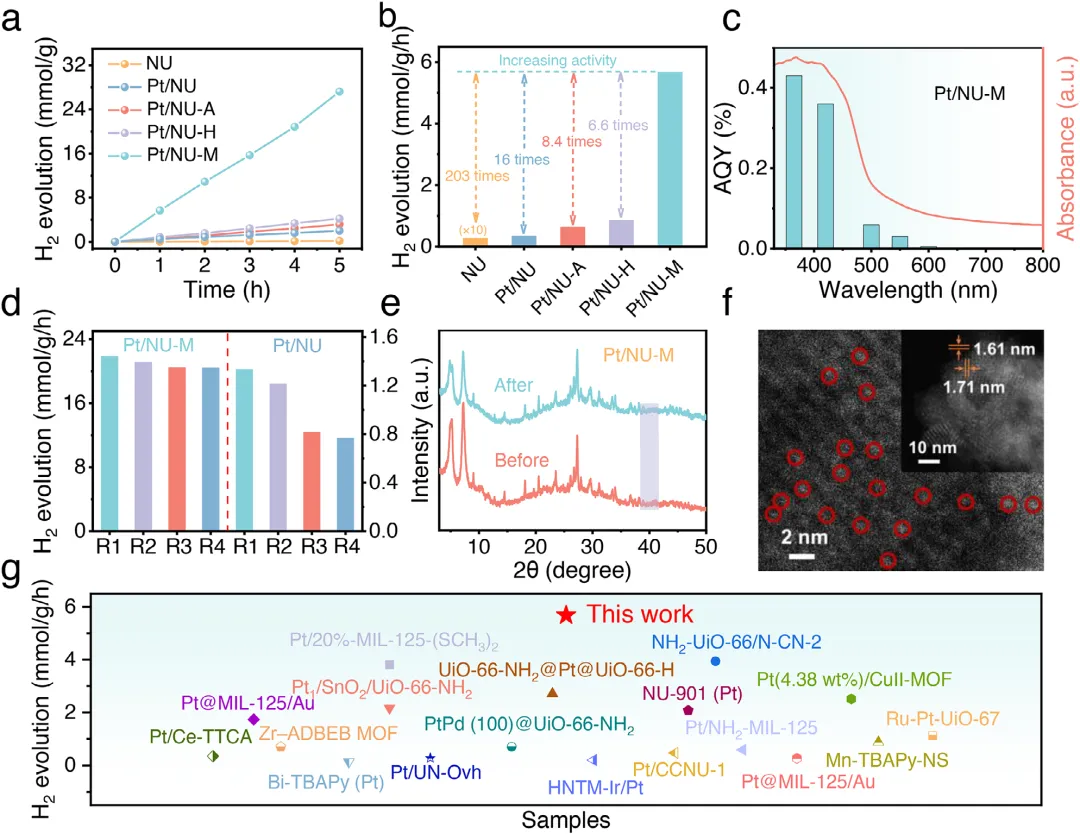
图1:Pt/NU-X的制备示意图及形貌表征。(a)展示了Pt/NU-X(X=A,H,M)的制备示意图。(b,c)分别为Pt/NU-A和Pt/NU-H的TEM图像,显示Pt纳米颗粒均匀分散。(d)为Pt/NU-M的AC HAADF-STEM图像,显示Pt原子级分散。(e,f)为Pt/NU-M的相应元素mapping,证实Pt单原子在催化剂中的均匀分布。图2:Pt/NU-X的结构与配位表征。(a)为NU、Pt/NU、Pt/NU-A、Pt/NU-H和Pt/NU-M的PXRD图谱。(b)为NU、Pt/NU、Pt/NU-A、Pt/NU-H和Pt/NU-M的FTIR光谱。(c)为NU-M和Pt/NU-M的S 2p XPS光谱。(d)为Pt/NU、Pt/NU-A、Pt/NU-H和Pt/NU-M的Pt 4f XPS光谱。(e)为Pt箔、PtS<sub>、PtO<sub>、Pt/NU、Pt/NU-A、Pt/NU-H和Pt/NU-M在Pt L<sub>边的归一化XANES光谱。(f-h)分别为Pt/NU-A、Pt/NU-H和Pt/NU-M的实验数据与拟合FT-EXAFS曲线对比。(i)为Pt/NU-A、Pt/NU-H和Pt/NU-M的L<sub>加权EXAFS数据小波变换。图3:Pt/NU-X的光学性质与电荷分离转移。(a)为NU、Pt/NU、Pt/NU-A、Pt/NU-H和Pt/NU-M的UV-vis DRS光谱。(b)为Pt/NU、Pt/NU-A、Pt/NU-H和Pt/NU-M的能带结构。(c)为NU、Pt/NU、Pt/NU-A、Pt/NU-H和Pt/NU-M的LSV曲线。(d)为NU、Pt/NU、Pt/NU-A、Pt/NU-H和Pt/NU-M的SPV光谱。(e)为NU、NU-M、Pt/NU和Pt/NU-M的瞬态光电流响应光谱。(f)为NU、NU-M、Pt/NU和Pt/NU-M的电化学阻抗谱。(g)为Pt/NU、Pt/NU-A、Pt/NU-H和Pt/NU-M的内建电场强度。(h)为NU、NU-M、Pt/NU和Pt/NU-M的稳态PL光谱。图4:原位表征与超快光谱。(a-c)分别为Pt/NU-A、Pt/NU-H和Pt/NU-M的原位XPS光谱。(d-f)分别为Pt/NU-A、Pt/NU-H和Pt/NU-M在400nm激发下于H<sub>O中的飞秒瞬态吸收光谱时间演化,监测440-700nm光谱窗口内8ns显示范围。下方面板为基于顺序模型全局分析获得的演化相关差谱(EADS)。图5:光催化性能与稳定性。(a)为NU、Pt/NU、Pt/NU-A、Pt/NU-H和Pt/NU-M的光催化产氢性能。(b)为NU、Pt/NU、Pt/NU-A、Pt/NU-H和Pt/NU-M使用抗坏血酸作为牺牲剂时的光催化产氢速率。(c)为Pt/NU-M光催化产氢的波长依赖AQY。(d)为Pt/NU和Pt/NU-M光催化产氢的稳定性测试。(e)为Pt/NU-M光催化产氢4循环前后的PXRD图谱。(f)为Pt/NU-M光催化产氢后的STEM图像(红圈代表Pt单原子)。(g)为Pt/NU-M与其他MOF基光催化剂的光催化产氢速率比较。本研究通过Lewis碱基团(-SH、-OH、-NH<sub>)功能化芘基MOFs,系统调控Pt助催化剂与MOFs之间的电子金属-载体相互作用(EMSI),实现了Pt从纳米颗粒到单原子的精确分散调控。其中,-SH配位诱导的最强EMSI使Pt以原子级分散状态稳定存在,形成Pt-O/S配位结构,同时建立强内建电场促进电荷分离转移,并通过Pt表面氧化降低反应能垒,最终实现了5.68 mmol g<sub><sup> h<sup>的优异光催化产氢速率(约为对照组16倍)和20小时以上的长期稳定性。该研究为通过分子级配位工程调控EMSI、设计高效单原子/纳米颗粒催化体系提供了普适性范式,在光催化制氢等清洁能源领域具有重要应用前景。Lewis-Base Coordination Enables Highly Dispersed Pt Cocatalyst on Pyrene-Based MOFs for Enhanced Photocatalytic Hydrogen Evolution,Angewandte Chemie International Edition,2026,https://doi.org/10.1002/anie.8241756