太原科技大学王洪雷/刘一鸣&上海交通大学曹红帅,最新AEM!富空位CoO/高自旋CoOOH异质界面双位点级联机制实现高效/超稳定HMF电氧化!
「上海岱算科技有限公司」已向境内外230余家高等院校/科研院所提供了累计1400多项模拟计算服务,赋能科学研究提速增效!合作实验课题组在线发表学术论文期刊有ACS系列、AM系列、Angew、CEJ、EST、JACS、Matter、Nature子刊等,助力科研工作锦上添花!5-羟甲基糠醛(HMF)电氧化为2,5-呋喃二甲酸(FDCA)为生物质增值制备可再生聚酯单体提供了一条可持续途径,然而在碱性条件(pH 11.0)下,非贵金属催化剂难以同时实现低过电位、高FDCA选择性以及超长稳定性。
2026年05月11日,太原科技大学刘一鸣、王洪雷团队在Advanced Energy Materials期刊发表题为“Dual-Site Cascade Mechanism Over Vacancy-Rich CoO/High-Spin CoOOH Heterointerfaces Enables Highly Efficient and Ultrastable 5-hydroxymethylfurfural Electrooxidation”的研究论文,王洪雷为论文第一作者,王洪雷、海南医科大学罗贤柱、西安近代化学研究所李冲、刘一鸣、上海交通大学曹红帅为论文共同通讯作者。
第一作者:王洪雷
通讯作者:王洪雷、罗贤柱、李冲、刘一鸣、曹红帅
通讯单位:太原科技大学、海南医科大学、西安近代化学研究所、上海交通大学
论文DOI:10.1002/aenm.70985
该研究中,密度泛函理论DFT计算揭示,富含氧空位的CoO显著降低了HMF到5-羟甲基-2-呋喃甲酸(HMFCA)转化的能垒,而低配位高自旋CoOOH有效稳定了*OH中间体以促进深度氧化。在这些理论见解的指导下,开发了一种紫外激光烧蚀结合电氧化活化策略,构建了富含氧空位的CoO/低配位高自旋CoOOH异质结构催化剂。该催化剂在仅1.19V(vs. RHE)的电位下即可达到10mA cm⁻²,实现了>98%的HMF转化率、近100%的FDCA选择性以及超过1000小时的稳定性,为同类条件下的非贵金属催化剂树立了新基准。实验及多种原位表征证实,反应路径的协同优化与理论预测高度吻合。这种氧空位诱导的低配位高自旋态调控策略表现出优异的普适性,成功扩展至镍基体系,并在多种生物质平台分子的电氧化中展现出显著提升的性能。该研究为设计用于生物质电增值的高效、稳定且成本低廉的非贵金属催化剂提供了新范式。
5-羟甲基糠醛(HMF)是一种重要的生物质衍生平台分子,可选择性氧化为2,5-呋喃二甲酸(FDCA),FDCA是石油基对苯二甲酸的理想可再生替代品,也是生产高性能聚呋喃二甲酸乙二醇酯(PEF)的关键单体。与传统的热催化氧化相比(通常需要高温、高压氧气和贵金属催化剂),电催化HMF氧化反应(HMFOR)在温和条件(室温和常压)下进行,在能效、环境友好性和与可再生电力的兼容性方面具有显著优势。该方法已成为一种极具前景的将生物质转化为高附加值化学品的策略。尽管近期取得了相当大的进展,但在碱性条件(pH 11.0)下同时实现高转化率、接近100%的FDCA选择性和超长期稳定性仍然是一项艰巨的挑战。虽然贵金属催化剂表现出优异的活性和选择性,但其高昂的成本阻碍了大规模应用。相比之下,非贵金属催化剂(尤其是钴基氧化物)通常在关键的决速步骤(包括C-H键活化和深度醛氧化)中动力学缓慢,并且在精确控制反应路径方面存在挑战。因此,通常需要高过电位(>1.4 V vs. RHE),表现出有限的长期耐久性(<500 h),并提供相对较低的底物转化率(<80%)。
先前研究表明,对钴基氧化物进行结构修饰(通过引入氧空位、调控局域配位环境或调节钴的自旋态(高自旋/低自旋)),可以显著优化反应物和关键中间体在活性位点的吸附/脱附行为,从而显著提高HMFOR的电催化动力学。然而,氧空位、配位几何构型和钴自旋态之间复杂的相互作用,以及它们对催化性能的协同效应,仍未得到充分阐明,这严重限制了对结构-性能关系和活性增强起源的更深入机理理解。此外,HMFOR涉及多步顺序的中间体转化,特别是在碱性条件(pH 11.0)下,关于合理的催化剂设计如何加速这些中间体的快速选择性转化的系统性研究,尤其是在原位和机理视角方面,明显缺乏。尽管最近的报道通过上述修饰策略展示了性能的提升,但对于特定结构基元如何控制反应路径并协同实现对多个中间体高选择性、近乎完全转化为FDCA的全面机理探索,在很大程度上仍是未解之谜。
在此,DFT计算首先揭示,富氧空位CoO显著降低了HMF到HMFCA转化的能垒,而低配位高自旋CoOOH稳定了Co-*OH构型,以促进HMFCA高选择性深度氧化为FDCA。在这些原子尺度的见解指导下,通过紫外激光烧蚀随后进行电化学氧化活化,开发了一种氧空位介导的CoO-高自旋CoOOH异质结催化剂。在碱性介质(pH 11.0)中,该催化剂在1.19 V vs. RHE的极低电位下驱动10 mA cm⁻²的电流密度,实现了>98%的HMF转化率、接近100%的FDCA选择性以及超过1000小时的卓越稳定性,在同等条件下超越了最先进的非贵金属钴基HMFOR电催化剂。原位拉曼光谱结合像差校正HAADF-STEM证实,富空位CoO在低电位下原位重构为低配位高自旋CoOOH。互补的原位光谱和液相色谱-质谱联用(LC-MS)分析阐明了富氧空位CoO与高自旋CoOOH之间的协同机制,证明了对多步HMF氧化路径的精确调控,与DFT预测高度吻合。此外,底物范围实验和普适性测试验证了这种氧空位诱导重构和高自旋态协同策略的鲁棒性和广泛适用性,为设计高效非贵金属电催化剂提供了一个合理的范式。
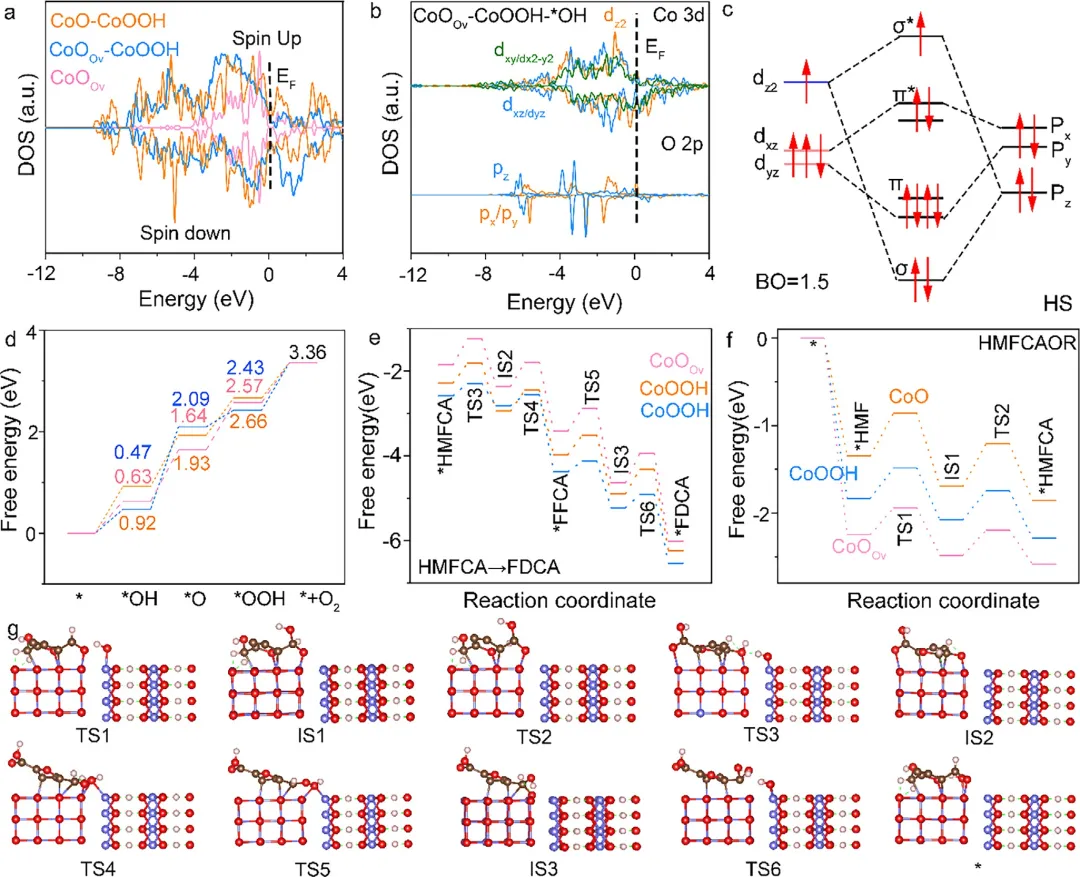
图1 | DFT计算。(a) CoOOv、CoO-CoOOH和CoOOv-CoOOH的PDOS。(b) Co 3d (CoOOH)和O 2p (*OH)的PDOS。(c) 自旋态调控示意图(HS态)。(d) CoOOv、CoO-CoOOH和CoOOv-CoOOH遵循AEM机制的OER自由能步骤图。(e) 在CoOOv-*OH、CoOOHLS-*OH和CoOOHHS-*OH模型上HMFCA氧化为FDCA的吉布斯自由能变化。(f) 在CoO、CoOOv和CoOOH模型上HMF氧化为HMFCA的吉布斯自由能变化。(g) CoOOv-CoOOH催化HMFCA各阶段对应的级联催化过程催化结构。
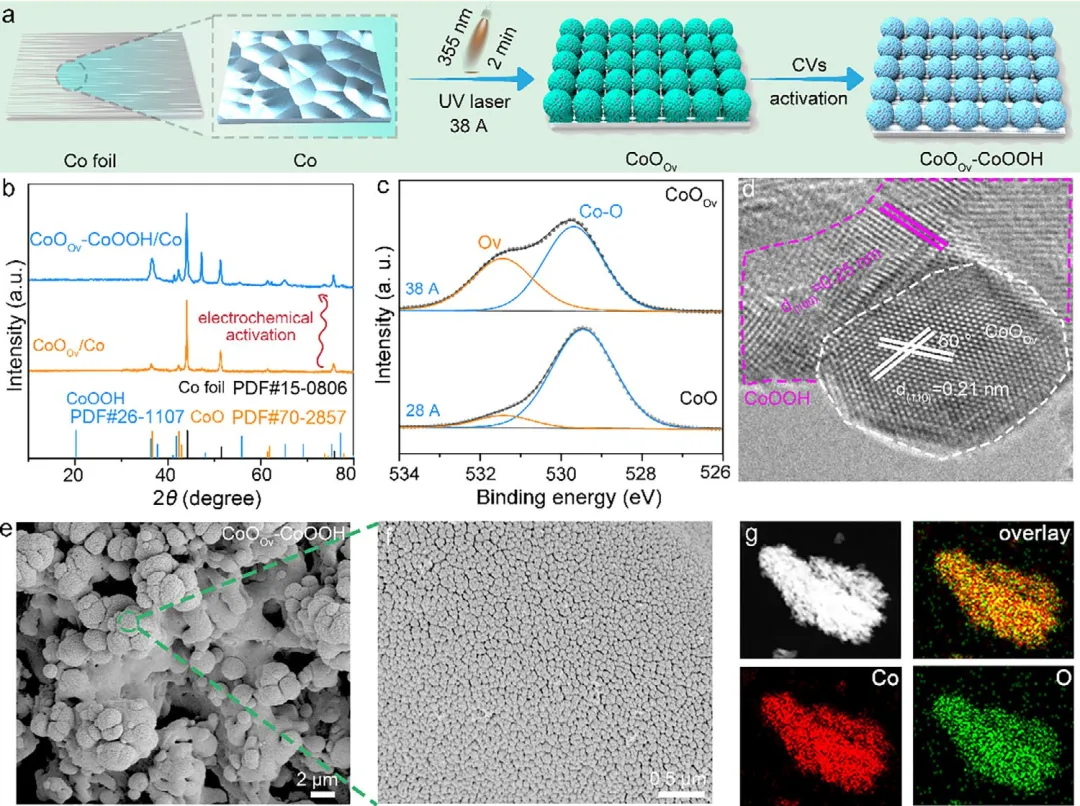
图2 | CoOOv-CoOOH异质结构催化剂的形貌与结构表征。(a) CoOOv和CoOOv-CoOOH的合成示意图。(b) 生长在钴箔上的CoOOv和CoOOv-CoOOH的XRD图谱。(c) 不同激光电流强度下制备的CoO样品的高分辨O 1s XPS谱图。(d) CoOOv-CoOOH的HAADF-STEM图像。(e,f) CoOOv-CoOOH的SEM图像。(g) CoOOv-CoOOH的STEM元素分布图(Co和O)。
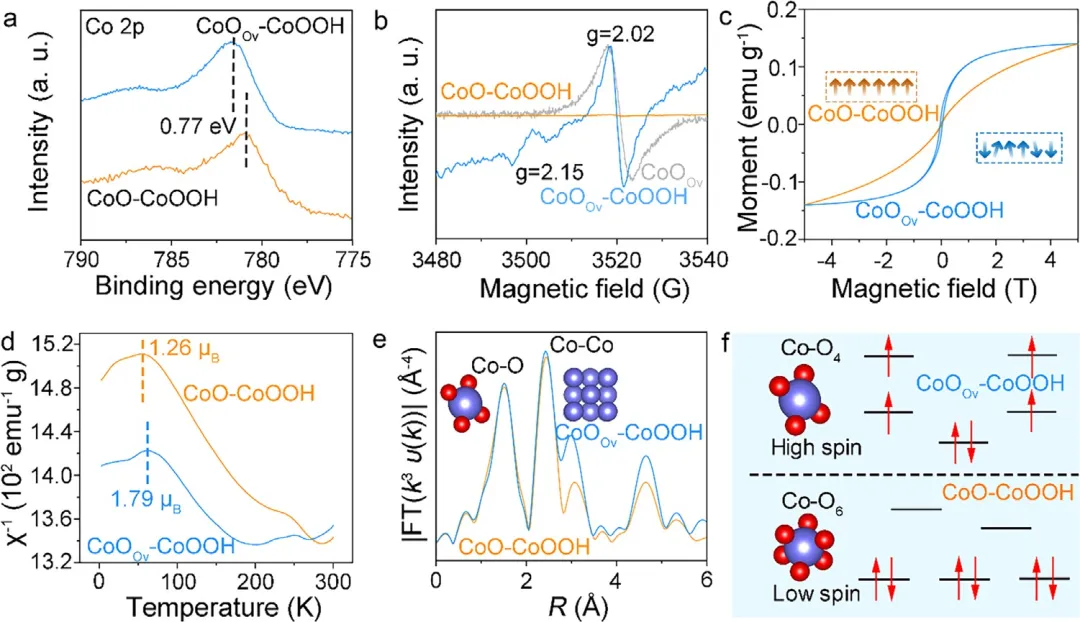
图3 | CoO-CoOOH和CoOOv-CoOOH中电子与局域原子结构的表征。(a) CoO-CoOOH和CoOOv-CoOOH的Co 2p核能级XPS谱图。(b) CoOv、CoO-CoOOH和CoOOv-CoOOH记录的电子顺磁共振(EPR)谱图。(c) 室温下测得的场依赖磁化强度。(d) CoO-CoOOH和CoOOv-CoOOH的饱和磁矩。(e) CoO-CoOOH和CoOOv-CoOOH的k³加权EXAFS振荡傅里叶变换到R空间图。(f) 结构模型和d轨道分裂示意图,说明低自旋(t₂g⁶)和高自旋(t₂g₄e_g²)Co³⁺构型。
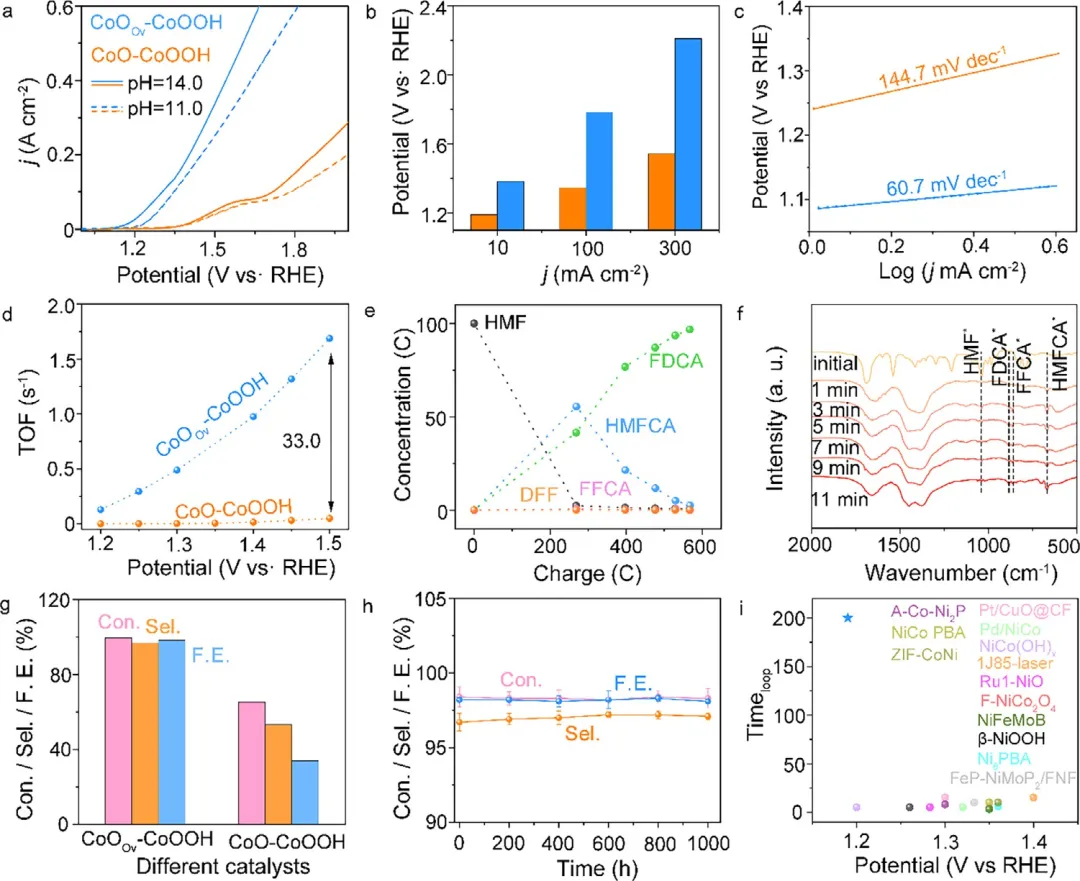
图4 在含有100 mM HMF的0.001 M KOH溶液中的电化学HMF催化性能。(a) CoO-CoOOH和CoOv-CoOOH在0.001 M KOH和1.0 M KOH中的极化曲线。在含有100 mM HMF的0.001 M KOH中:(b) 不同电流密度下的过电位值对比。(c) Tafel曲线。(d) TOF值。(e) CoOv-CoOOH催化HMFOR的色谱图。(f) 不同HMFOR时间的红外追踪光谱图。(g) CoO-CoOOH和CoOv-CoOOH的HMF转化率、FDCA选择性和法拉第效率。(h) CoOv-CoOOH在1.30 V vs. RHE下,于200个连续循环(每个循环5.0 h)中对100 mM HMF的HMF转化率、FDCA选择性和FDCA法拉第效率。(i) CoOv-CoOOH(0.001 M KOH)在10 mA cm⁻²下的稳定性和过电位与强碱性溶液(1.0 M KOH)中现有最先进电催化剂的对比。
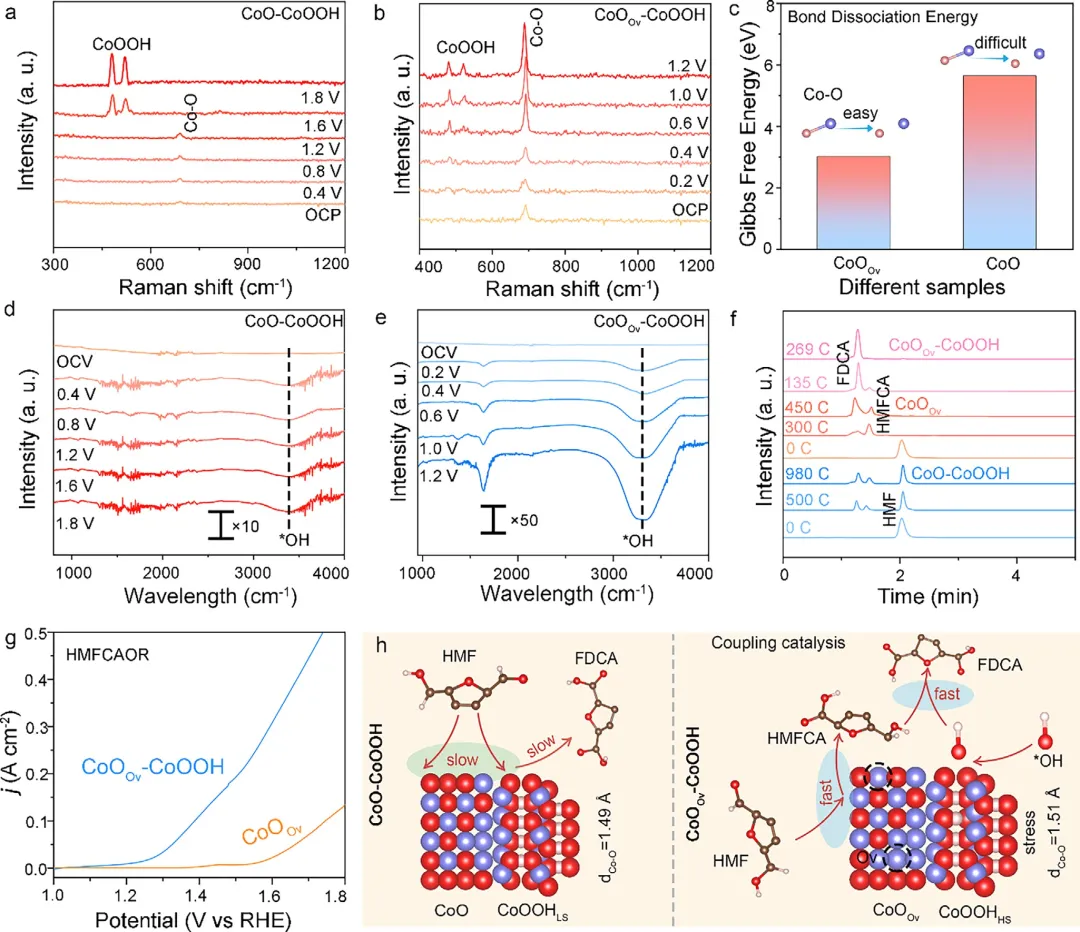
图5 | 支持HMFOR中级联催化形成的实验证据。(a, b) CoO–CoOOH (a) 和 CoOOv–CoOOH (b) 在100 mM HMF + 0.001 M KOH中,从开路电位(OCP)随电位增加至1.8 V (1.2 V vs. RHE) 记录的原位拉曼光谱。(c) CoO和CoOOv中Co–O键解离所需的吉布斯自由能。(d, e) HMFOR过程中CoO–CoOOH (d) 和 CoOOv–CoOOH (e) 的原位ATR-SEIRAS光谱。(f) 使用不同催化剂(CoO–CoOOH、CoOOv和CoOOv–CoOOH)进行HMFOR过程中,HMF、FDCA、HMFCA、FFCA和DFF浓度随通过电荷量的变化曲线。(g) CoOOv–CoOOH和CoOOv在含100 mM HMFCA的0.001 M KOH中的极化曲线。(h) 提出的CoO–CoOOH和CoOOv–CoOOH在HMFOR过程中的催化机理。
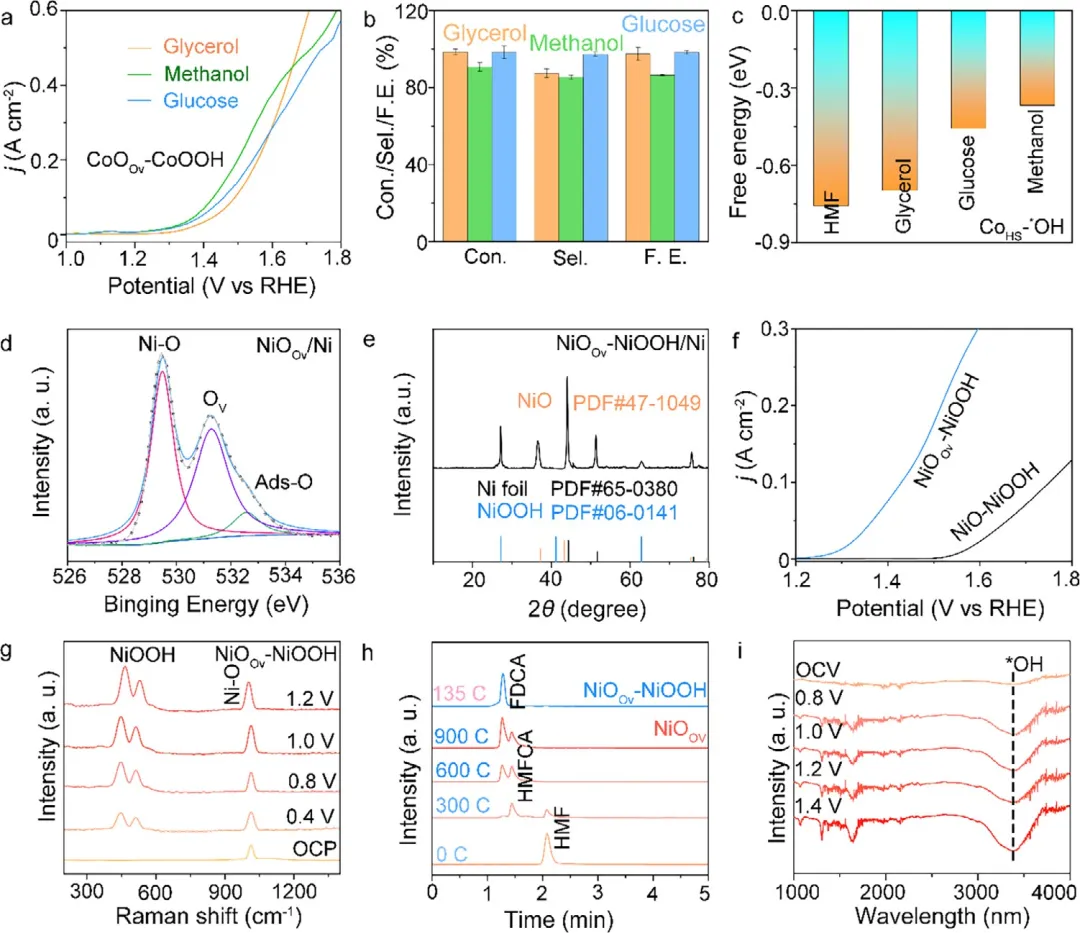
图6 | 级联催化普适性探索。(a) CoOOv-CoOOH催化剂在含100 mM甘油、甲醇和葡萄糖的0.001 M KOH中的LSV曲线。(b) CoOOv-CoOOH对甘油、葡萄糖和甲醇的转化率、选择性和法拉第效率。(c) CoHS-*OH中间体在HMF、甘油、葡萄糖和甲醇上吸附能力的比较。(d) NiO样品的高分辨O 1s XPS谱图。(e) 生长在钴箔上的NiOOv的XRD图谱。(f) NiOOv-NiOOH和NiO-NiOOH在0.001 M KOH中的极化曲线。(g) NiOOv-NiOOH在100 mM HMF + 0.001 M KOH中,从开路电位(OCP)随电位增加至1.2 V vs. RHE记录的原位拉曼光谱。(h) 使用不同催化剂(NiOOv和NiOOv-NiOOH)进行HMFOR过程中,HMF、FDCA、HMFCA、FFCA和DFF浓度随通过电荷量的变化曲线。(i) HMFOR过程中NiOOv-NiOOH的原位ATR-SEIRAS光谱。
总之,该研究合理设计了一种CoOOv-CoOOH异质结构,在碱性HMF电氧化(pH 11.0)中实现了超低过电位、近100%的FDCA选择性和>1000小时的稳定性,为非贵金属生物质电氧化催化剂树立了基准。原位光谱和DFT表明,氧空位驱动CoO重构,稳定高自旋Co³⁺,并实现了一条级联路径:CoOOv促进HMF → HMFCA,而高自旋Co³⁺诱导*OH加速HMFCA → FDCA,克服了经典的选择性-深度权衡。该策略具有广泛适用性,显著增强了镍基催化剂和其他生物质平台分子的电氧化性能。研究提出的“氧空位-高自旋级联催化”范式为设计用于可持续生物质升级的先进、低成本且耐用的电催化剂提供了一种通用且有效的方法。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
