
文章摘要
单重态(S1)和三重态(T1)激发是光催化过程中两条主要的、相互竞争的途径,在光催化过程中扮演着关键而又截然不同的角色。实现S1和T1激发能之间的灵活切换仍然是一个挑战。在该文章中,通过在骨架中集成折叠构建块,成功地合成了三种二维共价有机骨架(COF),它们的偏置堆积角分别为90°、105°和128°。结果表明,战略性偏置堆积可以有效地利用π-σ吸引,从而诱导从S1态到T1态的系间交叉。面对面堆积的BDT-HHTP-COF倾向于沿着电子转移途径,从而产生⋅O2−。相反,具有最佳偏置堆积距离的BDT-CTC-COF产生高浓度的1O2,这主要归因于能量转移途径。理论计算表明,BDT-CTC-COF能增强库仑相互作用,触发系间交叉,加速T1激子向骨架中吸附的O2的转移。机理途径中的这种转换是至关重要的,因为高度亲电的1O2在攻击甲苯的富含电子的芳香环方面显示出优越的效果,启动了选择性氧化过程,迅速实现了98%以上的降解率和80%的二氧化碳矿化,比以电子转移为主的途径提高了1.5倍。
背景介绍
利用太阳能驱动化学转化的光催化过程,基本上是由半导体中光生电子-空穴对的动力学决定的。电子转移(ET)和能量转移(ENT)是光催化过程中两条主要且经常相互竞争的途径,在光催化过程中扮演着关键而又截然不同的角色。由直接光子吸收产生的ET途径代表了传统的机制,它包括用于还原的电子和用于氧化的空穴的界面转移以启动氧化还原反应。这一过程促进了热力学上要求很高的氧化还原转化,并产生了高活性的中间体来推动有机合成。相反,ENT通过无辐射的偶极-偶极共振机制进行,其中被激发的催化剂将其激发能量作为一个整体传递给近端的受体分子,促进其进入更高的能量状态,而不需要净电子交换。此外,ENT途径在复杂基质中表现出高度的稳健性,因为其效率受到常见无机阴离子和溶解有机化合物的影响较小。通过绕过物理电荷迁移步骤,ENT还抑制了电荷载流子的复合,从而为利用光生激子提供了一条潜在的更有效的途径。因此,ENT作为一种复杂而强大的机制应运而生,它提供了优越的选择性、强大的抗干扰性和独特的激活通道,使其成为开发高性能光催化系统的关键战略。
作者提出了一种新的方法,通过精确调节2D COF的层间偏移π-π堆积来切换S1电子转移和T1能量转移路径。具体地说,通过在骨架中集成折叠构建块,成功地合成了三种基于噻吩衍生物的2D COF,它们的偏置堆叠角分别为90°、105°和128°。结果表明,战略性偏置堆积可以利用有效的π-σ吸引,从而诱导从S1态到T1态的系间交叉,从而产生三重态激子。面对面堆积的BDT-HHTP-COF光催化剂更容易发生S1电子转移途径,从而产生超氧自由基(⋅O2−)。相反,具有最佳偏置堆积距离的BDT-CTC-COF产生更高的单线态氧浓度(1O2),主要归因于T1能量转移途径。光物理表征和理论计算表明,BDT-CTC-COF能够增强库仑相互作用,触发系间交叉,加速T1激子向骨架中吸附的O2的转移。机理途径中的这一转换是至关重要的,因为高度亲电的1O2在攻击甲苯的富含电子的芳香环方面显示出优越的效果,启动了选择性氧化过程,从而迅速地获得了98%以上的降解率和80%的二氧化碳矿化效率,是电子转移为主的途径的1.5倍。显著的降解效率,再加上已阐明的以1O2为主导的非自由基氧化机理,突显了工程化COF结构在高级光催化环境修复中操纵能量转移途径的巨大潜力。
结果与讨论
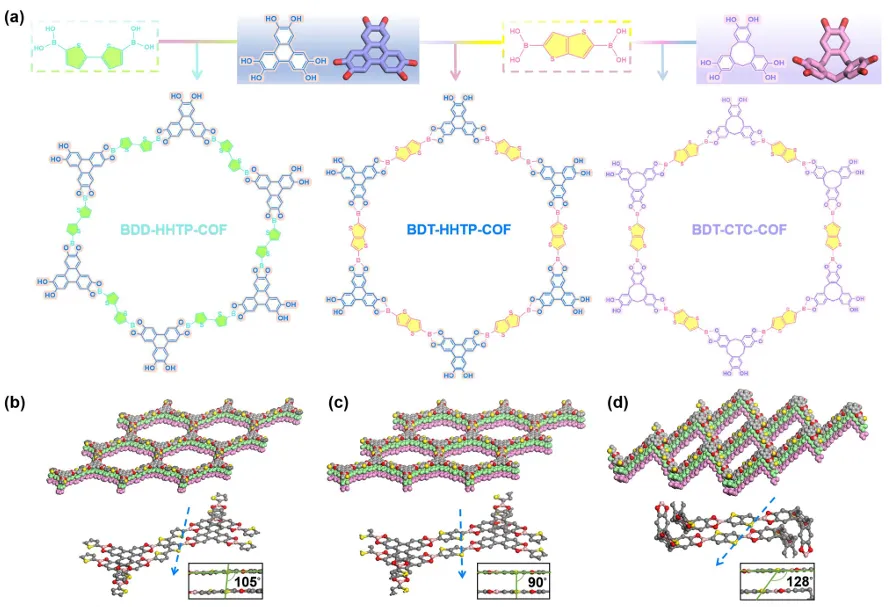
图1. (a) COF的合成路线和化学结构; (b) BDD-HHTP-COF、(c) BDT-HHTP-COF和(d) BDT-CTC-COF的空间填充模型。
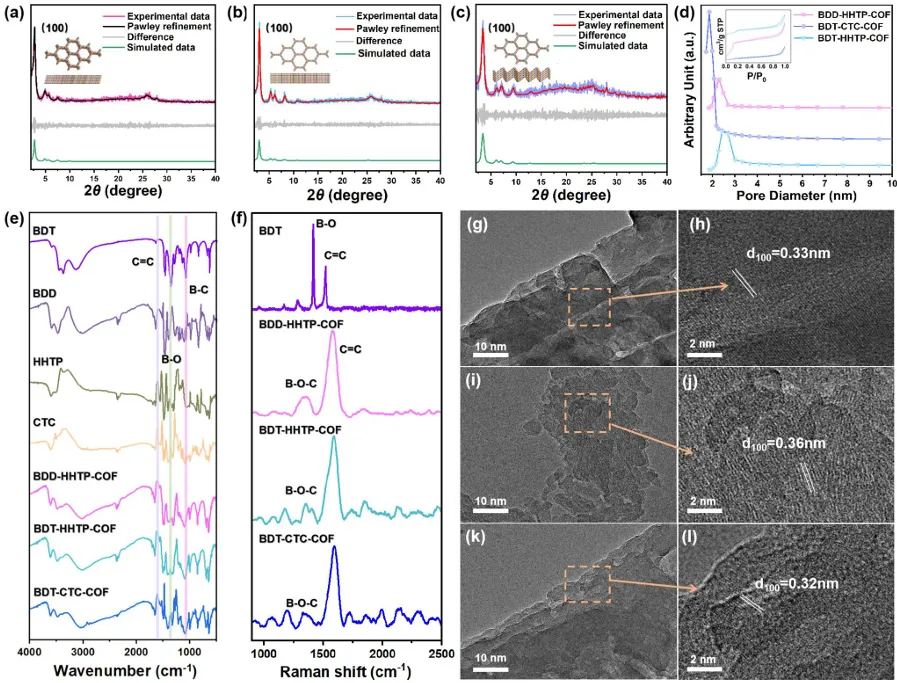
图2. (a) BDD-HHTP-COF、(b) BDT-HHTP-COF和(c) BDT-CTC-COF的实验衍射图和模拟衍射图; (d)碳纤维的氮吸附等温线和孔径分布; 单体和碳纤维的(e)FT-IR光谱和(f)拉曼光谱; (g-l)碳纤维的高分辨透射电子显微镜。
通过硼酸脱水和缩合将折叠构建块整合到骨架中,合成了三种具有层间面对面堆积或不同偏置堆积模式的2D COF(图1a)。利用Material Studio软件对合成的COF的详细结构信息进行了模拟。如图1b-d所示,由于构建块的立体化学配置不同,BDT-CTCCOF和BDD-HHTP-COF表现出不同程度的层间错位。用粉末X-射线衍射仪(PXRD)表征了这三种碳纤维的中等结晶结构(图2a-c)。这三个COF呈现出尖锐的衍射峰,显示出高质量的晶体结构。BDT-HHTP-COF在2.7、5.4、7.2、9.6和25.2°处出现衍射峰,分别对应于(100)、(110)、(210)、(220)和(001)面,与BDT-CTC-COF和BDD-HHTP-COF相似。用傅里叶变换红外光谱(FT-IR)表征了硼酸酯键的形成。在三个COF中观察到的1650 cm−1附近的伸缩振动峰来自苯环骨架中的C═C键。1375和1085 cm−1处的特征振动峰分别对应于硼酸酯单元中的B─O和B─C键。同时,与HHTP和BDT单元相比,BDT-HHTP-COF在3000-3600 cm─1的波数范围内表现出显著减弱的O─H信号(图2e)。在三种碳纤维的拉曼光谱中,1398 cm─1处的B−O伸缩振动消失,而1315 cm−1处出现一个新的宽峰,进一步证实了硼酸酯键的形成(图2f)。这些结果表明这三种COF光催化剂的合成是成功的。由于层间滑动将主要影响比表面积和孔大小,为了进一步验证可逆的结构转变,在77 K下通过氮气吸附测量了硼酸基COF的孔隙率(图2d)。计算得到BDT-CTC-COF、BDT-HHTP-COF和BDD-HHTP-COF的Brunauer-Emmet-Teller表面积分别为178.09、613.36和273.07 m2 g−1。平均孔径分布分别为1.86、2.48和2.29 nm。扫描电子显微镜(SEM)图像显示,由于层间堆积模式的不同,三种COF具有不同的形貌。BDT-HHTP-COF呈扁平片状,BDT-CTC-COF形态类似于由微米和纳米粒子组成的球形堆积,BDD-HHTP-COF呈花椰菜状。高分辨电子显微镜(HR-TEM)进一步证实了COFS的结构特征。可以清楚地观察到BDD-HHTP-COF、BDT-CTC-COF和BDT-HHTP-COF的晶格间距分别为3.30、3.60和3.20 Å(图g-l),这与模拟结果一致。
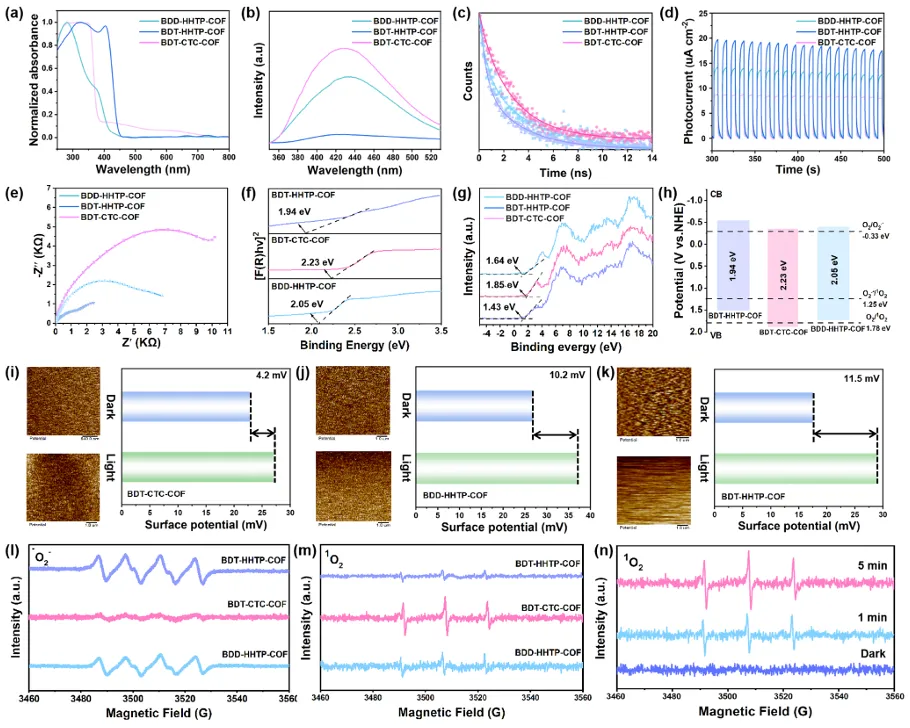
图3. (a) COF的UV-Vis DRS吸收光谱; (b) COF的光致发光光谱; (c) COF的TRPL衰减谱; (d) COF的瞬时光电流响应; (e) COF的环境影响报告书; (f) COF的Tauc图; (g) COF的XPS价带; (h) COF的电子能带结构示意图; (i-k) COF的KPFM表面电位图及光照前后相应的电势差; (l) DMPO存在下∙O2−的电子顺磁共振谱; (m-n)温度作用下1O2的EPR谱。
考察了所制备的三种碳纤维的光催化性能。首先,详细研究了层间堆积方式对激子过程的影响。UV-Vis漫反射光谱(UV-Vis DRS)表明,所制备的COF光催化剂在可见光范围内表现出最宽的吸附范围,这归因于最宽的共轭结构(图3a)。用光致发光光谱(PL)和时间分辨光致发光光谱(TRPL)研究了这些COF的激子效应(图3b,c)。BDT-CTC-COF具有最强的发光强度和最长的发光寿命,这可能是由于S1物种的ISC过程增强或电子-空穴对的复合所致。
用瞬时光电流和开尔文探针力显微镜(KPFM)研究了表面电荷分布(图3d,i-k)。BDT-HHTP-COF表现出最强的光电流密度和最大的表面电势差11.5 mV,表现出很强的光致电子-空穴分离效率。而BDT-CTC-COF具有最低的光电流密度和4.2 mV的表面电势差,这表明BDT-CTC-COF具有较小的内建电场强度,并抑制了激子解离成电子-空穴对,促进了激子的转移。电化学阻抗谱(EIS)也证实了BDT-HHTP-COF主导的ET途径和BDT-CTC-COF主导的ENT途径(图3e)。此外,还分析了作为半导体的重要因素的能隙(Eg)。根据Tauc图,计算出BDT-HHTP-COF、BDT-CTC-COF和BDD-HHTP-COF的Eg分别为1.94、2.23和2.05 eV(图3f)。根据价带X射线光电子能谱(VB-XPS)分析,BDT-HHTP-COF、BDT-CTC-COF和BDD-HHTP-COF的价带最大值(VBMs)分别为1.43、1.85和1.64 eV(图3g)。计算的导带极小值分别为−0.51、−0.38和−0.41 eV。这三种COF在热力学上都适用于各种氧还原反应,包括O2+e−→∙O2−,ENHE=−0.33 V(图3h)。然后,通过电子顺磁共振监测光催化反应中产生的∙O2−和1O2的活性物种,研究O2的活化途径。分别用TEMP和DMPO自旋捕获剂捕获1O2和∙O2−。可以清楚地看到,BDT-CTC-COF表现出最强的1O2信号,几乎检测不到∙O2−信号,而BDT-HHTP-COF表现出完全相反的模式。这表明BDT-CTC-COF通过ENT途径激活氧,而BDT-HHTP-COF遵循ET途径(图3l-m)。在BDT-HHTP-COF上产生的ROS包括∙O2−和1O2,进一步证明了可以通过调节COF的层间偏移堆积来实现电子转移和能量转移路径之间的切换。此外,在BDT-CTC-COF上产生的1O2随着反应时间的延长而逐渐增加,证实了其在光照下的持续产生和延长的寿命(图3n)。
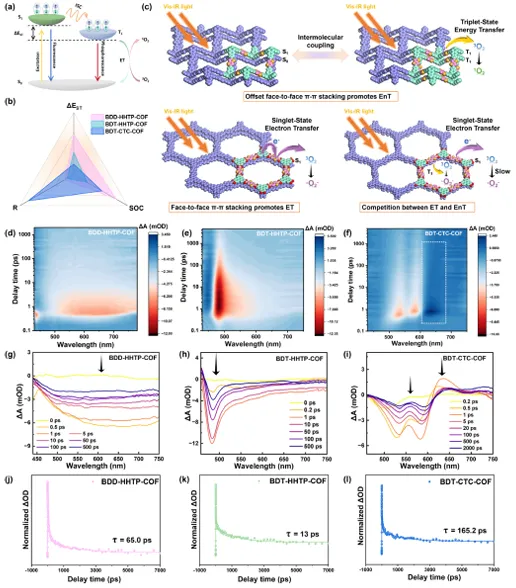
图4. (a)三重激子产生和1O2产生的可能的能量转移过程; (b)显示COF的主要光物理性质的雷达图; (c) COF光催化活化氧气的机理示意图; (d) BDT-HHTP-COF、(e) BDD-HHTP-COF和(f) BDT-CTC-COF; (g) BDT-HHTP-COF、(h) BDD-HHTP-COF和(i) BDT-CTC-COF的二维图; (j) BDT-HHTP-COF、(k) BDD-HHTP-COF和(l) BDT-CTC-COF的衰变动力学图。
通过密度泛函理论(DFT)计算揭示了相关的光激发过程的机制,以及COF的结构和激子行为之间的内在联系。根据以下公式计算SOC常量以获得ISC速率常数(KISC):
如图4a,b所示,BDT-CTC-COF表现出最大的R值和最小的ΔEST。较高的R值意味着自旋禁止的强烈驰豫和从S1到T1的快速转变,这对ISC过程是有利的。而在BDT-HHTP-COF上出现了完全相反的情况,这意味着ISC的过程受阻。BDD-HHTP-COF显示中间值,表示ET和ENT之间发生竞争。利用飞秒瞬时吸收光谱(FS-TAS)进一步揭示了激发态动力学的光物理行为。图4d-f显示了BDT-HHTP-COF、BDT-CTC-COF和BDD-HHTP-COF的二维伪彩色(ΔA)图随探测波长和延迟时间的变化。初始负信号主要是S1态的基态漂白(GSB),这可能归因于VB结构中电子的激发。正信号来自激发态吸收(ESA),激发态可以吸收基态无法吸收的光,从而过渡到更高的激发态。然后进行总体拟合分析,以阐明COF的激子跃迁漂白衰减动力学(图4g-i)。BDT-HHTP-COF在480 nm处显示出最强的GSB信号,表明其具有较高的激发态电荷密度。BDD-HHTP-COF更广泛的离域结构促进了电子的离域,从而产生了多个GSB信号。相反,BDT-CTC-COF在525-600 nm显示负的GSB信号,在600-675 nm显示正的ESA信号,对应于三重态激子吸收信号,随着时间的推移迅速衰减,证实了高效的ISC过程(图4j-l)。
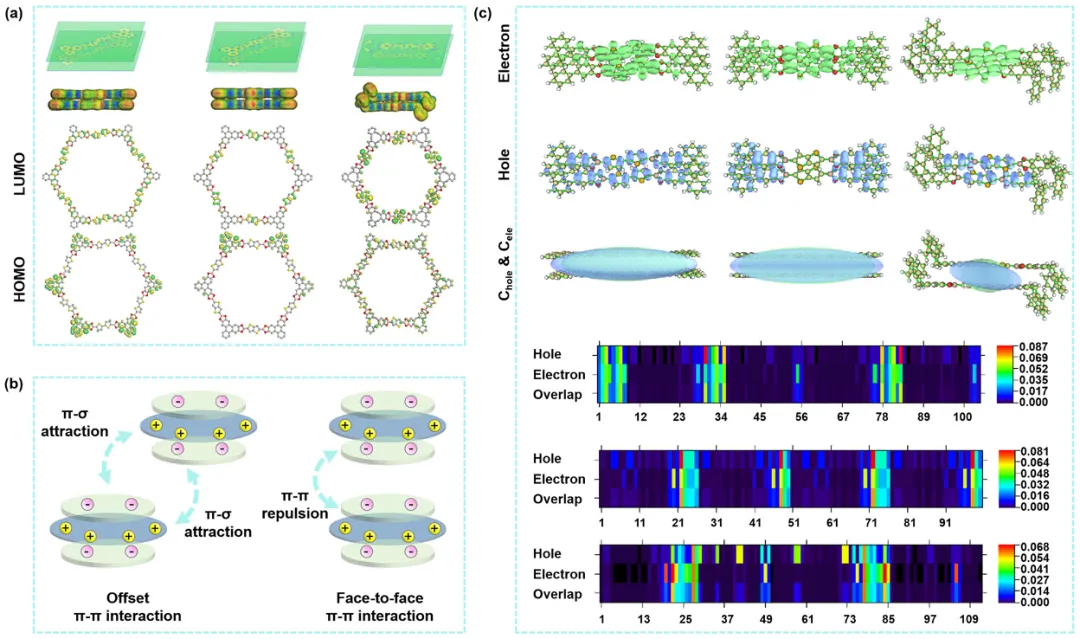
图5. (a) COF的电子密度分布和HOMO-LUMO; (b)芳香环之间的偏置和面对面堆积的不同静电相互作用图; (c)空穴(蓝)-电子(绿色)分布以及原子对空穴和电子的贡献。
通过理论计算进一步阐明了偏置堆积对激子跃迁过程的影响。分析了三个COF的最高占据分子轨道(HOMO)和最低未占据轨道(LUMO) (图5a)。HOMO主要位于苯环上,而LUMO主要位于噻吩衍生物上。在这些COF中,BDT-CTC-COF在HOMO和LUMO之间显示出最少的重叠。由于ΔEST仅产生于自旋相反的S1态价电子之间的电子排斥,偏移的π-π相互作用诱导了强烈的π-σ吸引,导致BDT-CTC-COF的ΔEST较低(图5b)。对主要激发态的空穴-电子分布分析发现,所有COF中的电子都明显局限在噻吩衍生物位点,涉及ORR(图5c)。BDT-HHTP-COF表现出最突出的电子-空穴分离效应。对于BDT-CTC-COF,偏移的π-π相互作用减弱了层间电子排斥作用。并且主要激发态中原子对空穴和电子的贡献也表明,噻吩衍生物位点处的电子分布更加明显,这与ORR的活性位点有着错综复杂的联系。
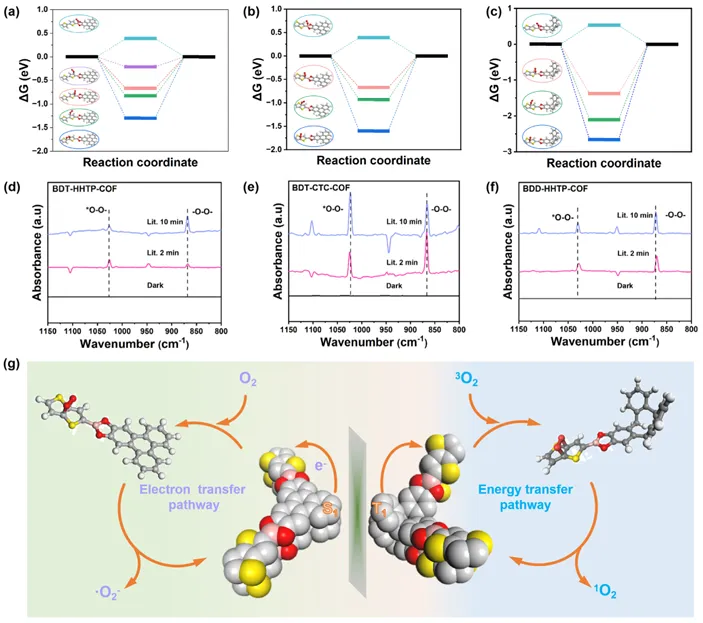
图6. (a-c)氧在不同COF位置的吉布斯自由能图; (d-f) COF光催化体系的原位漂移光谱; (g) BDT-CTC-COF中通过T1的能量转移和BDT-HHTP-COF中通过S1的电子转移的活化机理。
通过密度泛函理论计算,计算了这些光催化剂上不同位置的氧活化所需的吉布斯自由能。从图6a-c可以看出,这三个COF对O2活化的催化中心都在噻吩衍生物单元上,这些单元具有最低的吸附能。用原位漫反射红外傅里叶变换光谱(DRIFTS)研究了这些光催化剂上1O2和∙O2−的生成途径。图6d-f显示了三种催化剂在可见光照射下在高纯O2中随时间变化的漂移光谱。结果表明,在暗条件下峰值几乎没有起伏,而在光照条件下同时出现─O─O─和*─O─O─对应的860和1025 cm-1的特征峰。BDT-CTC-COF的*O─O─信号峰比─O─O─信号峰强,表明EnT介导的氧活化途径占主导地位。相反,BDT-HHTP-COF的─O─O─信号峰强于*─O─O─,证实了ET途径的主要作用。
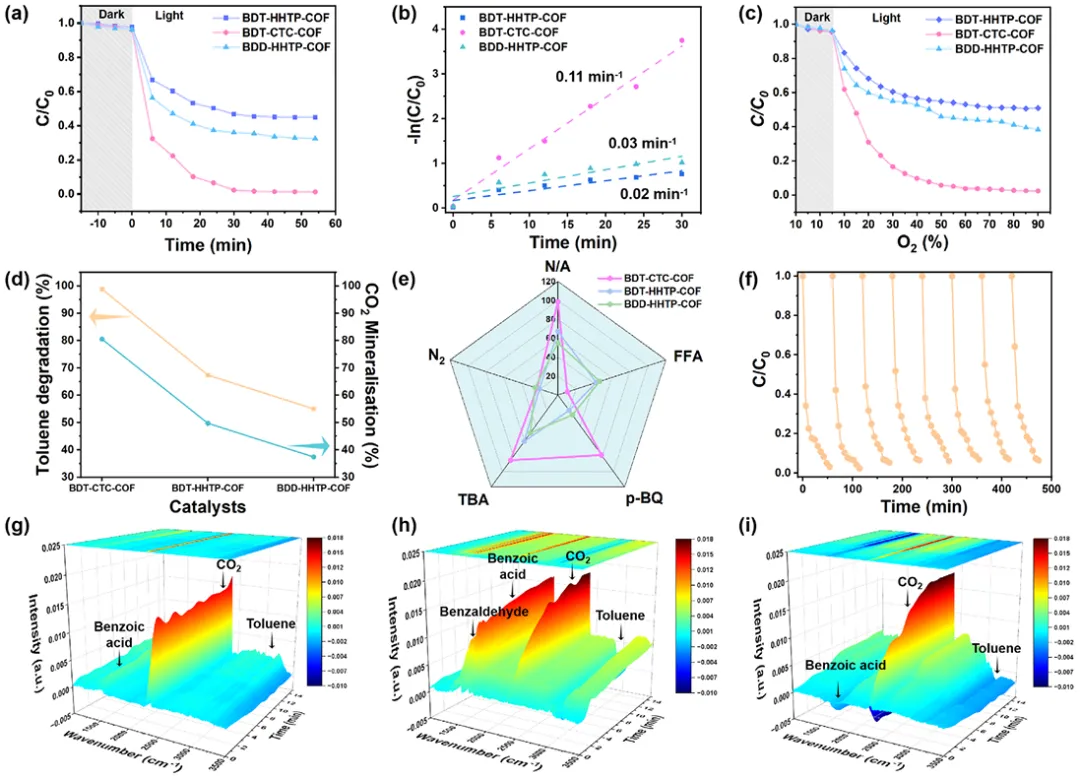
图7. (a)甲苯在COF上的降解曲线; (b) COF的拟一阶速率常数; (c)不同氧比下甲苯在COF上的降解曲线; (d)碳纤维的甲苯降解和二氧化碳矿化; (e)在不同清除剂的COF上光催化氧化反应中甲苯的降解; (f) COF的稳定性试验; (g-i)甲苯光催化氧化过程中碳纤维的原位红外光谱
甲苯对二氧化碳的矿化效率因所涉及的特定ROS而异。此外,COF的周期性孔结构和扩展的共轭骨架促进了甲苯分子的扩散和吸附。因此,以甲苯氧化为模型反应,评价了三种COF上氧气的光活化性能。在进行光催化实验之前,确保催化剂对甲苯分子处于吸附-脱附平衡状态,以区分吸附和光催化降解对甲苯的去除性能。然后在黑暗条件下将系统稳定15分钟。如图7a所示,BDT-CTC-COF的光催化活性最好,在30分钟内达到98.68%,其次是BDD-HHTP-COF,而BDT-HHTP-COF的降解效率最低。用Langmuir–Hinshelwood model (−ln(C/C0)=kt)计算了降解速率常数。特别是,BDT-CTC-COF代表了最佳的光降解速率常数(0.11 min−1),分别是BDD-HHTP-COF和BDT-HHTP-COF的3.8倍和5倍(图7b)。结果表明,1O2在光催化降解甲苯方面表现出优于∙O2−的性能。同样,对于不同氧气比例下甲苯的降解,BDT-CTC-COF具有最高的氧气利用率和最高的降解效率(图7c)。图7d显示,BDT-CTC-COF的CO2矿化率超过80%,分别是BDT-HHTP-COF和BDD-HHTP-COF的1.5倍和2倍。为了进一步验证甲苯降解过程中涉及的主要ROS,使用了各种清除剂作为捕捉剂来确定ROS的类型(图7e)。使用自由基清除剂,如呋喃醇、叔丁醇、对苯二酚(p-BQ)和鼓泡氮气,考察了1O2、羟基自由基(⋅OH)、∙O2−和光生电子对这些体系的影响。结果表明:BDT-CTC-COF体系中,1O2是主要的活性物种,对甲苯的降解有贡献;而在BDT-HHTP-COF体系中,∙O2−是主要的活性物种。值得注意的是,BDT-CTC-COF上甲苯的降解率在重复循环10次后仍保持在95%以上,这表明BDT-CTC-COF光催化剂能够在长期运行中保持高活性和稳定性(图7f)。此外,光催化测试后BDT-CTC-COF的X射线衍射谱、FT-IR光谱、BET分析和扫描电子显微镜图像也证实了其结构的稳定性。不同中间体在催化剂表面堆积的差异表明了分子氧在气固界面上活化的动力学差异。图7g-i显示了氧化过程中中间体强度随时间延长的变化。可以明显看出,由于甲苯的连续降解,苯甲酸和苯甲醛在BDD-HHTP-COF上的积累和苯甲酸在BDT-HHTP-COF上的积累持续增加,而BDT-CTC-COF上的积累几乎只表现为CO2的增加。
文章总结
综上所述,该文提出了一种通过精确调节层间偏移量π-π堆积来切换S1电子转移和T1能量转移途径的新策略。具体地说,通过在骨架内集成折叠构建块,成功地合成了三种不同偏移堆叠模式的2D COF。结果表明,战略性偏置堆叠可以利用有效的π-σ吸引,从而将ISC从S1态诱导到T1态。面对面堆积的BDT-HHTP-COF光催化剂更倾向于S1电子转移途径,从而产生∙O2−。相比之下,具有最佳偏移堆叠距离的BDT-CTCCOF产生更高的1O2浓度,这主要归因于T1能量转移途径。理论研究表明,BDT-CTC-COF可以增强库仑相互作用,促进ISC,并推进T1激子向O2吸附中心的转移,共同促进1O2光合作用的能量转移过程。此外,由于层间的偏移堆积,更多的活性中心可以暴露出来,进一步提高了光催化性能。机理途径中的这种转换是至关重要的,因为高度亲电的1O2在攻击甲苯的富含电子的芳香环方面表现出优越的效果,启动了选择性氧化过程,导致迅速达到98%以上的降解率和80%的二氧化碳矿化效率。阐明了S1和T1之间的转化机制,加上显著的光合作用效率,突显了工程化COF结构在高级光催化环境修复中的巨大潜力。
文章信息
Deng, Y.; Li, D.-K.; Luo, Y.-L.; Li, P.-F.; Xia, Z.-N.; Ci, P.; Bian, R.-J.; Gao, R.-Y.; Wu, X. Switching Between Singlet and Triplet Excitation in Covalent Organic Frameworks for Highly Efficient Photocatalysis. Angew. Chem. Int. Ed, 2026, e8843840.
文案:张瑞婷

