电化学 CO₂还原为乙烯等高值化学品是实现碳中和的重要途径,但 C-C 键形成的高能垒与析氢副反应竞争严重限制了多碳产物的选择性与效率。铜基材料是唯一可实现 CO 深度还原为烃类的催化剂,但传统铜催化剂结构不明确导致反应路径与机制模糊,理性设计高效铜基催化剂促进 C-C 偶联仍是该领域的核心挑战。金属有机框架的明确晶态结构为构建原子级精准活性位点提供了理想平台,N-杂环卡宾配体的强 σ 给电子能力可有效调控铜中心电子结构,但将卡宾铜位点与铜纳米颗粒整合实现协同催化的策略尚未得到探索。
太原理工大学张献明团队开发了后合成修饰构建卡宾铜与铜纳米颗粒协同活性位点的策略,实现了 CO₂电还原高选择性产乙烯,系统揭示了界面微环境与电子效应的协同调控机制。相关成果发表于《Angewandte Chemie International Edition》。
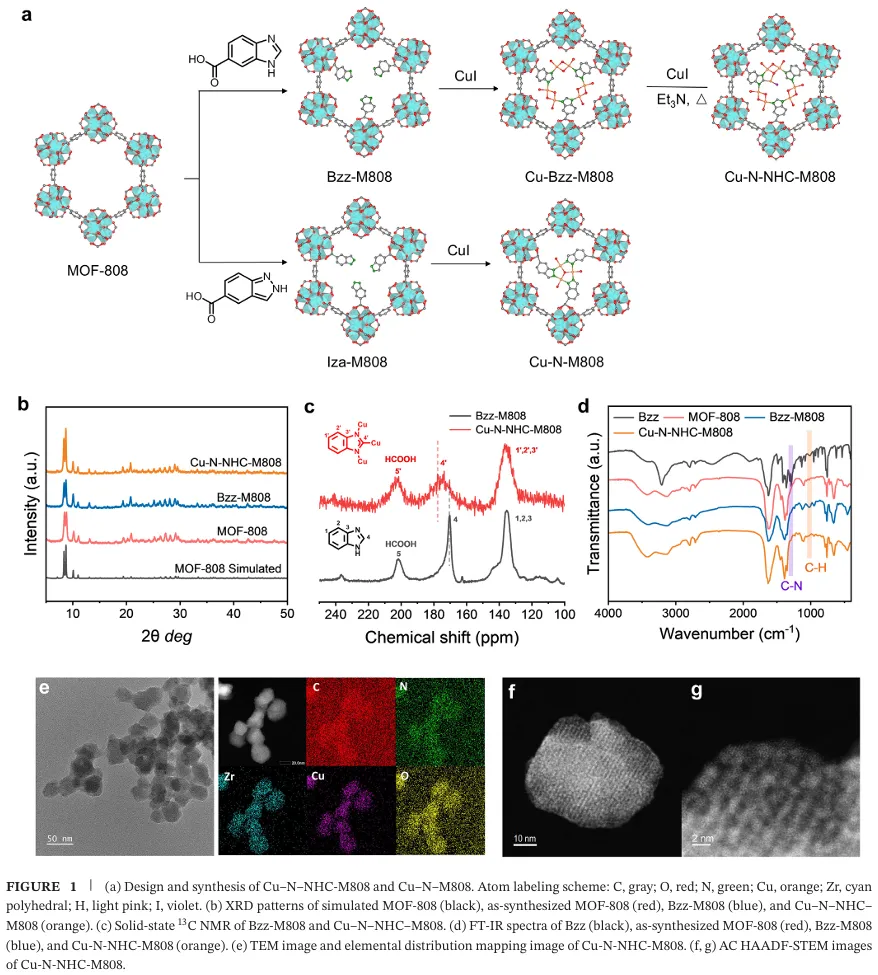
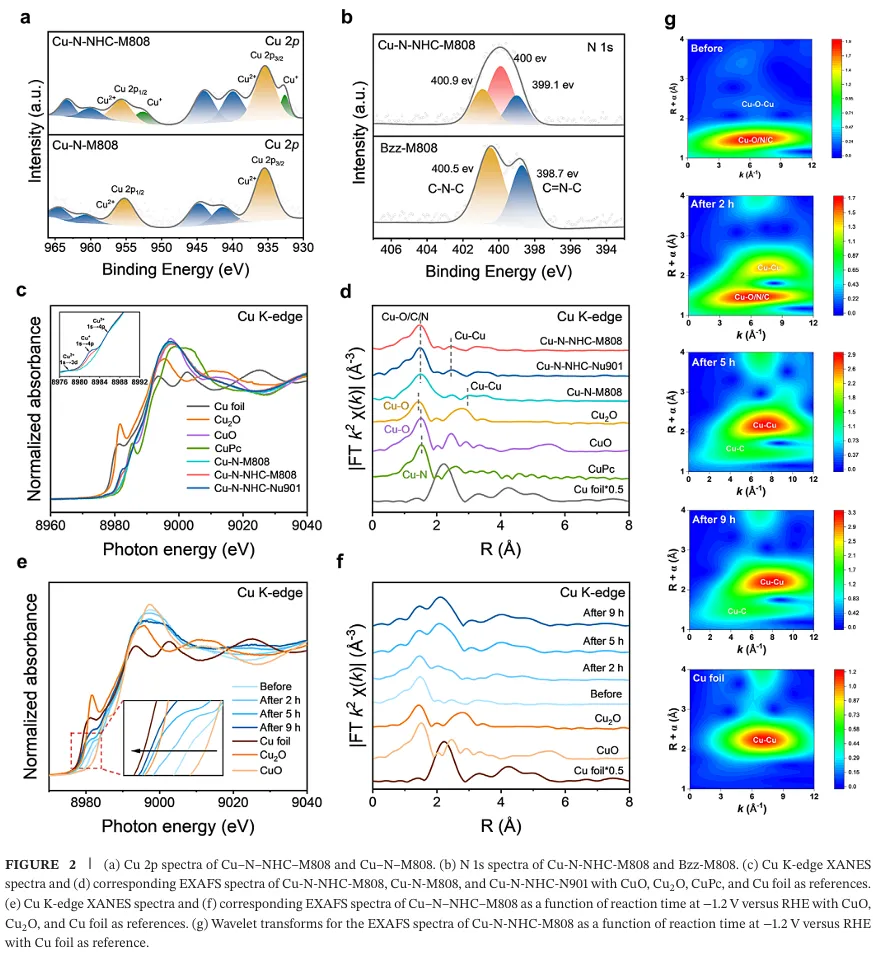
研究团队通过后合成修饰策略,在 MOF-808 框架中同时引入双 (μ-O) 双核铜位点与 N-杂环卡宾锚定的 Cu(I) 原子,构建 Cu-N-NHC-M808 催化剂(图 1)。粉末 X 射线衍射、固体核磁、红外光谱、电镜与 X 射线光电子能谱共同证实了配体接枝与卡宾铜活性位点的成功构建,X 射线吸收光谱验证了铜的混合价态与局域配位环境(图 2)。
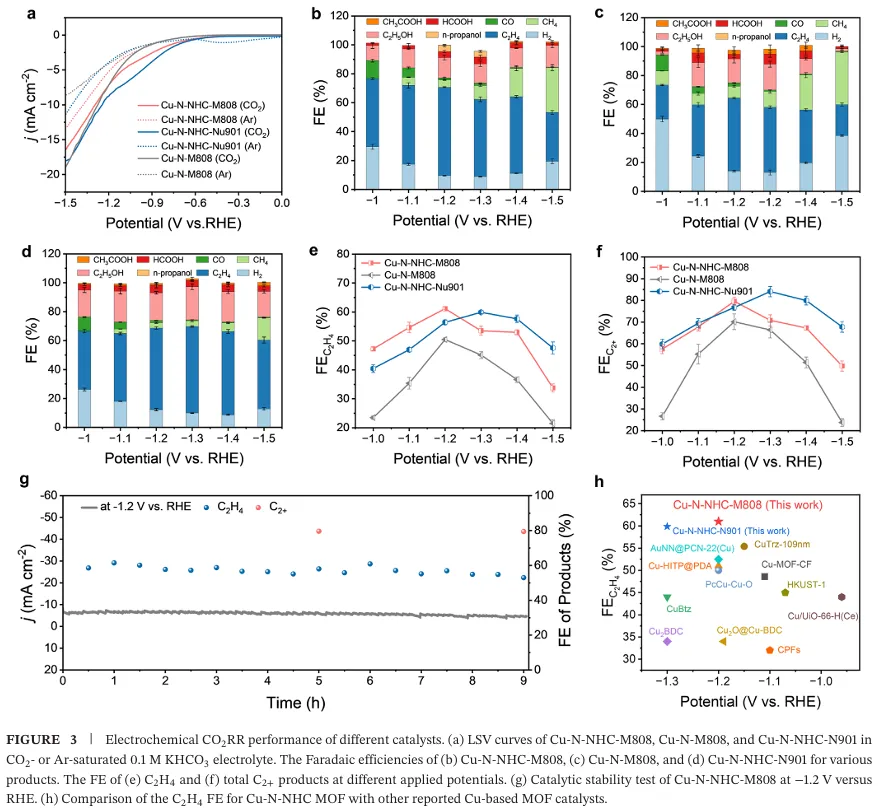
电化学性能测试显示,该催化剂在中性电解质中-1.2 V vs RHE 下实现 61.0% 的乙烯法拉第效率与 79.5% 的总 C₂产物法拉第效率,析氢反应被有效抑制(法拉第效率 < 10%),9 小时稳定性测试仍保持 52% 以上的乙烯选择性,性能显著优于无卡宾位点的对照催化剂与已报道的多数铜基 MOF 催化剂(图 3)。原位表征与理论计算共同揭示了催化机制,电解过程中双核铜位点原位重构为铜纳米颗粒,与稳定的卡宾铜位点形成协同活性中心(图 2)。
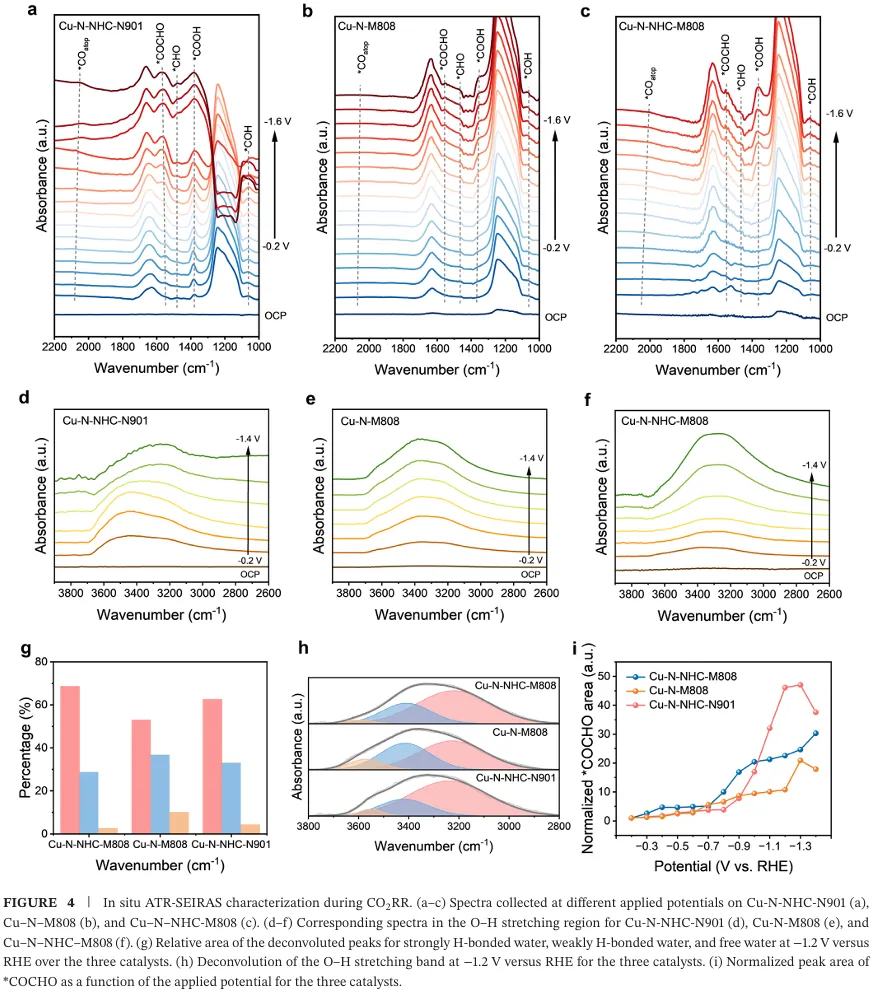
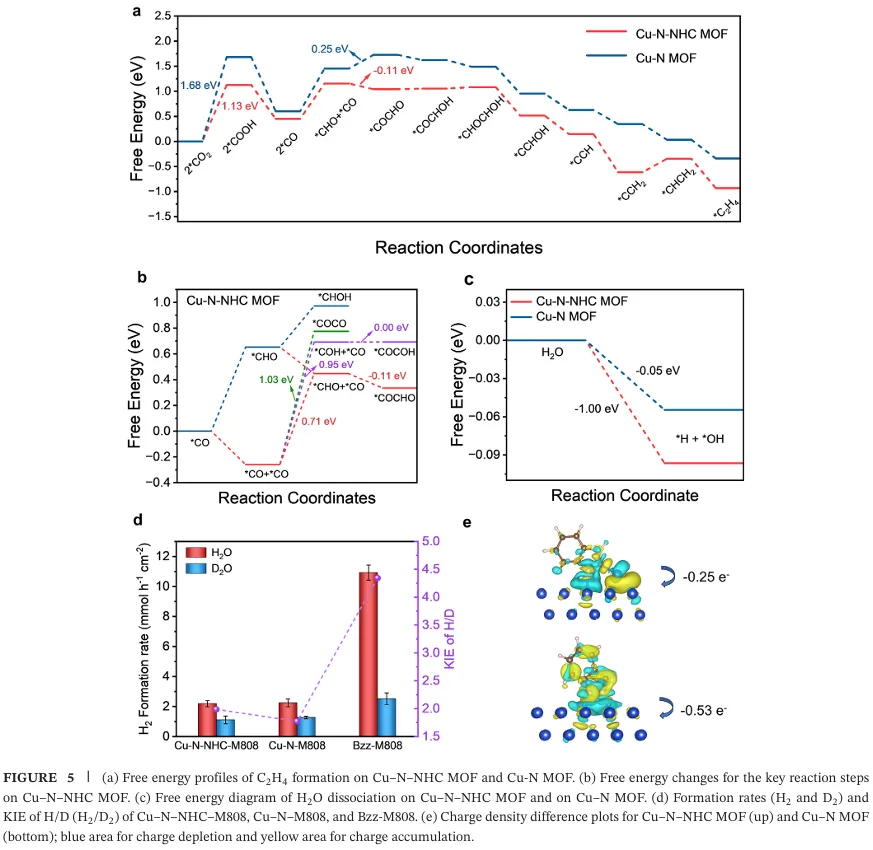
原位衰减全反射红外光谱证实卡宾配体促进界面水形成强氢键网络,加速质子转移与*CO 加氢生*CHO,动力学同位素效应实验验证了水解离与质子转移的促进作用(图 4)。密度泛函理论计算显示,卡宾铜位点将*CO 加氢能垒从 1.00 eV 降至 0.71 eV,不对称*CO-*CHO 偶联能垒低至-0.11 eV,强给电子特性增强关键中间体的吸附稳定,协同作用显著降低 C-C 偶联能垒并抑制析氢副反应(图 5)。
这项研究构建了 MOF-808 中 N-杂环卡宾锚定 Cu (I) 原子与原位生成铜纳米颗粒的协同催化体系,实现了中性电解质中 CO₂还原高选择性产乙烯。卡宾配体通过构建界面氢键网络促进质子转移,强给电子特性稳定关键反应中间体,分子位点与纳米颗粒的协同作用显著降低不对称 C-C 偶联能垒并抑制析氢副反应。该工作建立了金属有机框架中分子-纳米结构协同活性位点的设计策略,为高效电催化 CO₂转化催化剂的理性设计提供了全新思路。

L.-H. Jia and and X.-M. Zhang, Angewandte Chemie International Edition (2026): e7002606, https://doi.org/10.1002/anie.7002606.
本文仅用于学术分享。若有侵权,请联系我们,我们将及时修改或删除!





 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
