ZrO2不同结构形貌创制及热催化CO2/CH3OH直接合成DMC
刘乐1,王晓洋1,官修帅1,张小超1,2,3,4,张国强1,2,胡胜勇3,杨颂1,4,刘守军4
1太原理工大学化学与化工学院,山西 太原 030024;2茅台学院资源与环境学院,贵州 仁怀 564507;3太原理工大学安全与应急管理工程学院,山西 太原 030024;4太原科瑞康洁净能源有限公司,山西 太原 030024
刘乐, 王晓洋, 官修帅, 等. ZrO2不同结构形貌创制及热催化CO2/CH3OH直接合成DMC[J]. 化工进展, 2026, 45(4): 2308-2317.
>>DOI:10.16085/j.issn.1000-6613.2025-0730
利用CO2合成碳酸二甲酯(DMC)对推动可持续发展和实施“碳达峰、碳中和”战略具有重要的理论与现实意义。目前,CO2/CH3OH直接合成DMC在催化剂表面的演变过程尚不明晰,从而制约了高效催化剂的设计。本文通过调控Zr源和表面活性剂合成了具有不同形貌结构(花状、颗粒状、球形)的四方相ZrO2催化剂,并结合理论计算与原位表征系统探究了ZrO2表面催化CO2/CH3OH合成DMC的反应机理。性能测试结果表明,相比于花状和颗粒状ZrO2,球形ZrO2展现优异的DMC产率(5.11mmol/g)。透射电镜(TEM)表征证实球形ZrO2高度暴露的(101)活性晶面,促进了其对CO2的有效吸附-活化;BET测试显示,球形ZrO2具有更大的比表面积(67.8m2/g),从而提供更多CO2和CH3OH吸附活化位点;X射线光电子能谱(XPS)和电子顺磁共振(EPR)表征结果证实,球形ZrO2表面存在更多氧空位,增强了CO2有效活化;CO2/NH3-程序升温脱附(TPD)结果表明,球形ZrO2具备多种酸碱位点,可减少副反应的发生。原位红外光谱表征分析表明,CO2和CH3OH在球形ZrO2表面主要以双齿碳酸盐及单齿CH3O*物种,有利于关键中间体甲基碳酸酯的形成。理论计算表明,氧空位的存在降低控速步骤能垒(*CH3OCOO+*CH3→DMC,305.6kJ/mol→292.0kJ/mol),从而提高DMC产率。这一工作将为实现CO2转化为高附加值长链化学品提供新的思路和见解。
CO2是温室气体的主要组分且在碳资源循环利用过程中具备可持续性和丰富性。目前,研究人员致力于CO2转化为高附加值领域化学品,其中碳酸二甲酯(DMC)被誉为当今“绿色”有机化合成的新基石,由CO2/CH3OH直接合成DMC过程简单且绿色环保。在反应过程中,CO2先与一分子甲醇形成甲基碳酸酯中间体,脱除一分子水,再与一分子甲醇反应生成DMC。然而,反应受热力学限制和CO2难以活化等问题,导致DMC收率较低。那么,寻找一类高效活化CO2和提高DMC产率的催化剂是核心问题。
研究表明,ZrO2有3种晶相:立方相(c-ZrO2)、四方相(t-ZrO2)、单斜相(m-ZrO2),ZrO2晶相物化属性具有较强差异性。例如,Wang等通过理论计算研究了CO2/CH3OH在ZrO2的3种晶相表面合成DMC的反应机理,发现t-ZrO2具有最佳的催化活性。Jung等对比研究分析四方相和单斜相ZrO2上甲氧基和碳酸单甲酯的形成速率,发现t-ZrO2具有较高的酸碱位点密度和强度,使得四方相更容易形成DMC合成过程的重要中间体甲基碳酸酯。Tomishige等通过在不同温度下焙烧ZrO2·xH2O得到t-ZrO2和m-ZrO2催化剂,并对其进行NH3/CO2-程序升温脱附(TPD)测试。结果表明,t-ZrO2催化剂表面的酸性和碱性都较强,t-ZrO2更有利于合成DMC。理论计算和实验结果均表明,t-ZrO2更有利于合成DMC。目前,关于晶相的调控方法主要有负载金属、热处理、添加助剂、添加外场、形貌调控等。研究表明,固体催化剂的形态结构影响暴露的晶面、表面组成和微观结构,催化剂的形貌调控已被证明是调节活性位点及其在多相催化中催化性能的有效策略。
基于此,本文采用水热法制备球形、颗粒状和花状3种形貌的四方相ZrO2催化剂,探讨了其在CO2和甲醇合成DMC中的催化活性和反应机理。采用X射线衍射(XRD)、扫描电镜(SEM)、透射电镜(TEM)、NH3/CO2-TPD、X射线光电子能谱(XPS)、电子顺磁共振(EPR)和傅里叶变换红外光谱(FTIR)等一系列表征技术对催化剂的形貌特征和结构性质进行研究,探究了ZrO2形貌、暴露晶面、酸碱位点和氧空位浓度对催化CO2和甲醇直接合成DMC反应性能的影响规律,为CO2绿色转化为高附加值DMC提供良好的实验数据与科学依据。
球形ZrO2催化剂制备。首先,将1.387g硝酸氧锆和0.0346g十二烷基硫酸钠(SDS)溶解于30mL甲醇溶液中,标记为A溶液。其次,将1.802g的尿素溶解在30mL的甲醇溶液中,标记为B溶液。在持续机械搅拌下,将B溶液缓慢滴加到A溶液中,室温反应2h,得到C溶液。然后,将C溶液转移到100mL高压反应釜中,在120℃下水热反应24h。反应结束后,自然冷却至室温,通过离心机7000r/min下分离收集产物,并依次以蒸馏水和无水乙醇为溶剂依次洗涤两次。随后,将得到的样品在60℃恒温烘箱中干燥12h。最后,将样品在马弗炉中500℃煅烧5h,得到球形ZrO2催化剂标记为ZrO2-S。
颗粒状ZrO2催化剂制备。颗粒状ZrO2催化剂的制备方法与球形ZrO2的制备相似,将0.0346g的SDS替换为0.0427g的十六烷基三甲基溴化铵(CTAB),其他制备过程不变,便可得到颗粒状ZrO2催化剂,颗粒状ZrO2标记为ZrO2-P。
花状ZrO2催化剂制备。首先,将1.777g的Zr(SO4)2·4H2O和0.2953g的CH3COONa溶解到60mL离子水中,形成无色溶液。之后,将溶液转移至100mL聚四氟乙烯内衬不锈钢高压釜中,加热至200℃并在该温度下保持6h。然后,将高压釜自然冷却至室温,离心收集白色沉淀,用去离子水洗涤3次,在60℃下真空干燥12h,得到前体。最后,将前体在坩埚中600℃下以15℃/min的加热速率进行煅烧,形成白色粉末,制备的花状ZrO2标记为ZrO2-F。
采用X射线衍射仪(Shimadzu,XRD-6000 X)对催化剂的晶体结构进行表征,扫描范围为10°~80°,扫描速度为10(°)/min。使用扫描电子显微镜(Bruker XFlash 6130)以及透射电子显微镜(Thermo Fisher Scientific,TF20)进行形态和微观结构研究。样品的比表面积和孔径分布通过Brunaurer-Emmett-Teller(BET)和BarrettJoyner-Halenda(BJH)方法根据N2吸附-解吸等温线计算。采用电子顺磁共振光谱仪(Bruker EPR300E)检测催化剂中的氧空位含量。测量CO2/NH3-TPD以研究催化剂表面酸/碱度。红外(IR)测量是在配备漫反射傅里叶变换光谱(DRIFTS)反应器的光谱仪(Thermo,Nicolet 6700)上按照以下程序进行:①预处理,将10mg催化剂粉末放入原位池中,在氩气氛围下将催化剂在200℃下加热10min,用于除去部分预吸附的水和CO2;②CO2预吸附,在红外池中通入氩气(氩气流量为20mL/min),待气流稳定后,流动气体切换为CO2,流量为5mL/min;③甲醇吸附,在红外池中通入氩气,使氩气以5mL/min流量通过甲醇饱和器,并将甲醇带入红外池中。
DMC的合成在高压催化反应釜(100mL,西安太康生物仪器科技有限公司)中进行。一般情况下,在反应器中加入0.05g催化剂(ZrO2)和10mL甲醇。反应前,先用CO2气体(0.5MPa)冲洗反应釜3次以除去釜中空气,在室温下用CO2加压至1.5MPa。将反应釜升温至170℃,待反应釜冷却到室温,收集液相产物,将1-丙醇(CH3CH2CH2OH,99.8%)作为内标物。采用气相色谱仪进行分析。在本工作中,使用式(1)计算DMC产率。
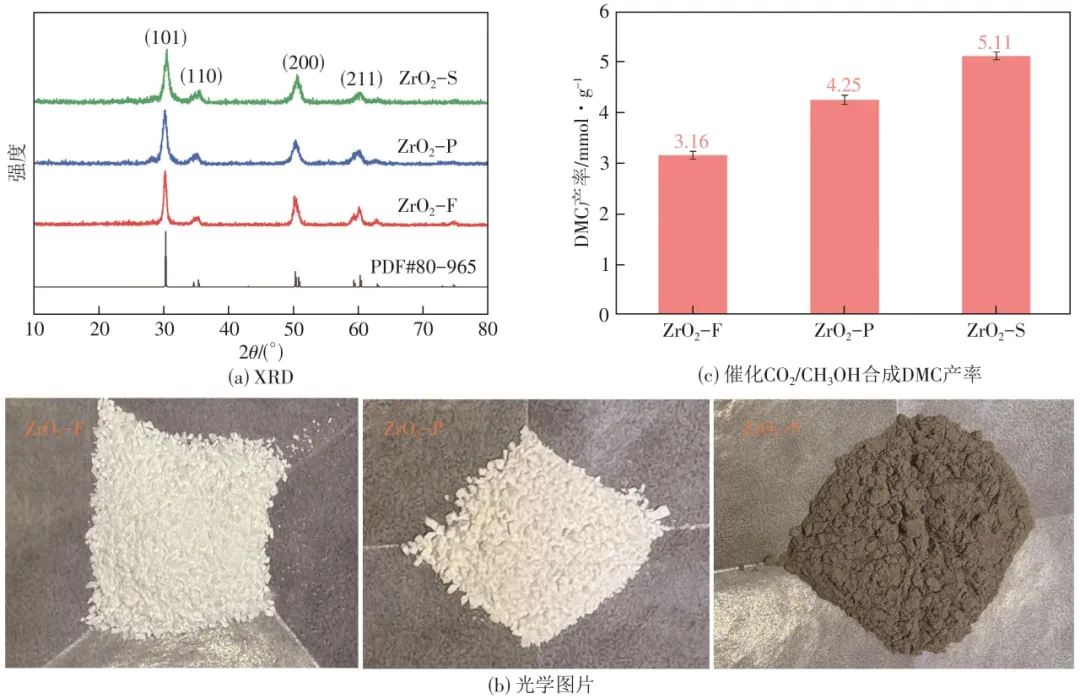
调控Zr源和表面活性剂制备了具有不同形貌结构(花状、颗粒状、球形)的四方相ZrO2催化剂,研究结构形貌对催化CO2/CH3OH合成DMC的影响。图1(a)显示了球形(ZrO2-S)、颗粒状(ZrO2-P)和花状(ZrO2-F)ZrO2催化剂的XRD图谱,用X射线衍射分析了晶体结构、衍射面和峰宽等。ZrO2的特征峰出现在30.2°、35.3°、50.8°和60.2°处,可分别归属于(101)、(110)、(200)和(211)晶面,所制备的ZrO2催化剂的特征峰对应于ZrO2标准卡(PDF#80-965),并未观察到其他衍射峰,表明无杂质相存在。ZrO2-F催化剂表现出银白色,CTAB表面活性剂改性的ZrO2-P呈现出灰白色,SDS表面活性剂改性的ZrO2-S相较于ZrO2-P颜色加深,呈现灰黑色,催化剂的这种颜色变化,说明催化剂表面物理化学性质发生了改变,见图1(b)。本文对热催化CO2/CH3OH合成DMC反应条件进行了考察,最佳反应条件为:反应时间3h、反应温度170℃、催化剂用量0.05g、CO2初始压力1.5MPa。ZrO2形貌差异体现出不同的催化活性,ZrO2-S、ZrO2-P和ZrO2-F催化剂在最佳反应条件下的DMC产率分别为5.11mmol/g、4.25mmol/g和3.16mmol/g,催化活性顺序为ZrO2-S>ZrO2-P>ZrO2-F,见图1(c),催化剂的这种活性差异可能与其表面物理化学性质有关。
图1 ZrO2-S、ZrO2-P和ZrO2-F催化剂的XRD图、光学图片和催化CO2/CH3OH合成DMC产率
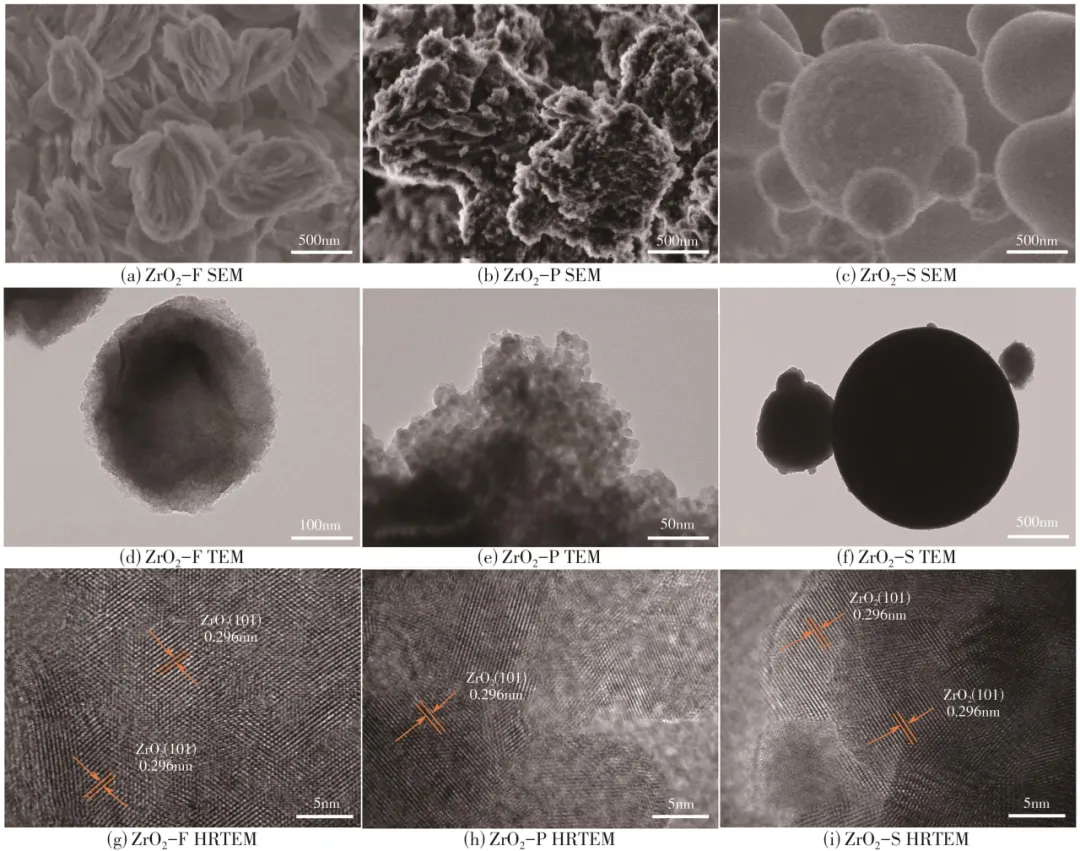
对ZrO2-S、ZrO2-P和ZrO2-F催化剂的形貌结构进行了研究分析,见图2(a)~(f)。ZrO2-F是由直径为300~500nm的纳米片堆叠成的花状结构,ZrO2-P由5~12nm的规则颗粒组成,ZrO2-S则由1~2µm的实心球体构成。ZrO2样品均显示规则的花状、颗粒状和球状形貌。通过高分辨率透射电镜(HRTEM)对ZrO2-S、ZrO2-P和ZrO2-F催化剂的分析可知,如图2(g)~(i)所示,ZrO2-S、ZrO2-P和ZrO2-F的晶面间距为0.296nm,对应四方相ZrO2的(101)晶面,ZrO2-S、ZrO2-P和ZrO2-F均主要暴露(101)晶面,表明四方相ZrO2制备成功。
图2 花状、颗粒、球形ZrO2的SEM、TEM和HRTEM图
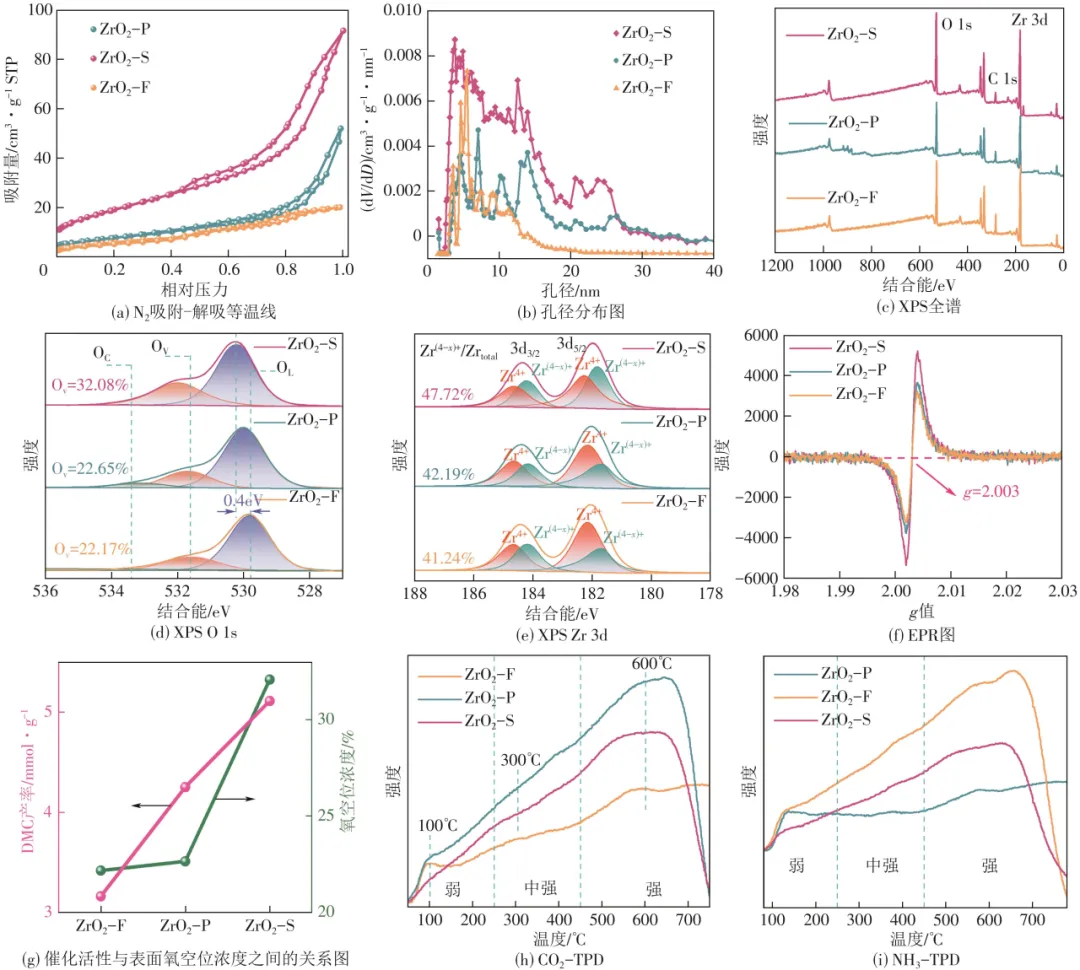
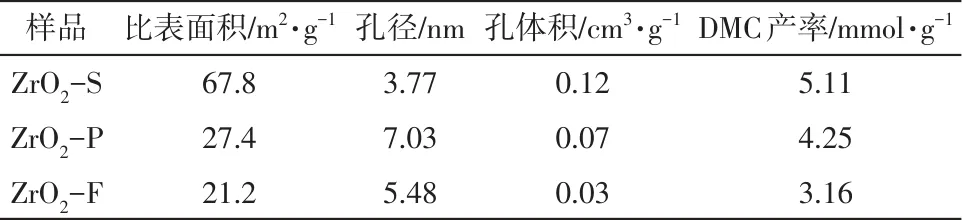
图3(a)、(b)为3种形貌ZrO2催化剂的N2吸附-脱附等温线和孔径分布图,可以看出3个样品吸附-脱附等温线均无明显的饱和吸附平台,且呈现Ⅳ型吸附-脱附等温线和H3型回滞环,表明3个样品都具有典型的介孔结构。不同条件下催化剂的比表面积、孔径、孔体积以及DMC产率列于表1。结果显示,ZrO2-S催化剂(67.8m2/g)比其他样品具有更高的比表面积,归因于ZrO2-S大量介孔(2~50nm)结构存在,为催化CO2/CH3OH提供了更多的活性位点。因此,具有较大比表面积和大量介孔结构的ZrO2-S催化剂(5.11mmol/g)表现出优于其他样品的DMC产率。
图3 ZrO2-S、ZrO2-P和ZrO2-F催化剂的表面性质
不同形貌ZrO2催化剂的元素组成和化学态已采用X射线光电子能谱进行了比较分析,见图3(c)、(d)。图3(c)所示样品XPS全谱图在531.45eV、285.14eV和183.48eV处分别对应于O 1s、C 1s和Zr 3d的峰,表明ZrO2的成功制备。XPS中O 1s区域可用于研究表面氧物种分布,见图3(d)。ZrO2-P和ZrO2-F在529.8eV、531.7eV和533.4eV处呈现出3个特征峰,分别归属于ZrO2中的晶格氧(OL)、氧空位(OV)和表面吸附氧(OC)。然而,ZrO2-S中的晶格氧、氧空位和表面吸附氧的峰位置向较高结合能处移动,在530.2eV、532.0eV和533.7eV处呈现3个特征峰,拟合后的峰位置向较高结合能处移动可归因于ZrO2-S中较高的OV浓度以及催化剂中氧环境发生的剧烈变化。由XPS结果可知,催化剂中OV浓度占比可通过OV/(OC+OV+OL)的积分峰面积表示。ZrO2-S、ZrO2-P和ZrO2-F的OV浓度分别为32.08%、22.65%和22.17%,催化剂中OV浓度顺序为:ZrO2-S>ZrO2-P>ZrO2-F。ZrO2-S中OV浓度明显高于ZrO2-P和ZrO2-F,表明ZrO2-S中更多氧空位存在。OV是还原金属氧化物中的常见缺陷,可以显著影响其表面性质,增强金属与载体相互作用,从而提高分散金属物质的稳定性。研究表明,材料的Zr 3d XPS谱图也可用于分析OV浓度,见图3(e)。ZrO2-S、ZrO2-P和ZrO2-F催化剂的中心峰在182.2eV和184.4eV处,分别归属于ZrO2的Zr 3d5/2和Zr 3d3/2。Zr 3d5/2和Zr 3d3/2在182.1eV、181.6eV、184.6eV、184.2eV处出现4个特征峰:184.6eV和182.1eV处归属于Zr4+,184.2eV和181.6eV处归属于Zr(4-x)+,氧化锆中Zr4+和Zr(4-x)+的比例与催化剂中OV浓度有关。使用Zr(4-x)+/[Zr(4-x)++Zr4+]表示催化剂中OV浓度,ZrO2-S、ZrO2-P和ZrO2-F的OV浓度分别为47.72%、42.19%和41.24%,催化剂中OV浓度顺序为:ZrO2-S>ZrO2-P>ZrO2-F。这与XPS O 1s谱图分析结果一致。
为了进一步研究样品的OV浓度,对样品进行了电子顺磁共振(EPR)测试,见图3(f)。对于不同形貌ZrO2催化剂,EPR光谱在g=2.003处的各向同性信号对应于具有俘获电子能力的氧空位的形成。EPR信号的强度与自由电子密度成正比,表明了氧空位的形成。ZrO2-S相较于ZrO2-P和ZrO2-F,表现出更高的衍射峰强度,然而ZrO2-P和ZrO2-F中衍射峰强度差别不大,表明ZrO2-S催化剂上具有更多的表面OV,其结果与XPS测试中O 1s和Zr 3d的分析结果保持一致。基于不同形貌ZrO2催化剂上OV浓度的巨大差异,本文对催化剂表面氧空位浓度与催化剂活性的关系进行了研究,见图3(g)。ZrO2-S、ZrO2-P和ZrO2-F的DMC产率分别为5.11mmol/g、4.25mmol/g和3.16mmol/g,DMC产率顺序为:ZrO2-S>ZrO2-P>ZrO2-F。这与XPS和EPR谱图中测得的OV浓度顺序保持一致。ZrO2催化CO2/CH3OH直接合成DMC产率随着OV浓度的增大而增大,具有最大OV浓度的ZrO2-S催化剂表现出最高的DMC产率(5.11mmol/g),DMC产率与催化剂表面OV浓度正相关。
据报道,表面OV可以作为路易斯碱性位点,在CO2的吸附-活化中起到关键作用。因此,本文通过CO2/NH3-TPD进一步研究了催化剂表面的酸碱性质。在50~250℃、250~450℃和>450℃温度范围内出现3个脱附峰,分别对应催化剂中弱酸碱性、中强酸碱性和强酸碱性,见图3(h)~(i)。催化剂的碱性位点越多,CO2的解吸温度越高。ZrO2-S、ZrO2-P和ZrO2-F在50~250℃都有一个明显的脱附峰(100℃),表示CO2与表面的碱性羟基发生反应,生成碳酸氢盐物种。ZrO2-S在250~450℃之间有一个小峰(300℃),表示CO2与碱性氧阴离子发生反应,生成双齿碳酸盐物种。ZrO2-S、ZrO2-P和ZrO2-F在450~800℃之间有一个较大的宽峰(600℃),表示CO2与晶格氧阴离子发生反应,生成单齿碳酸盐物种。ZrO2-S在100℃、300℃和600℃具有多个峰,表明催化剂表面存在多种碱性位点。ZrO2-S的多种碱性位点有望促进CO2的吸附-活化。同时,ZrO2-S的多种酸性位点有利于甲醇活化形成CH3*物种。
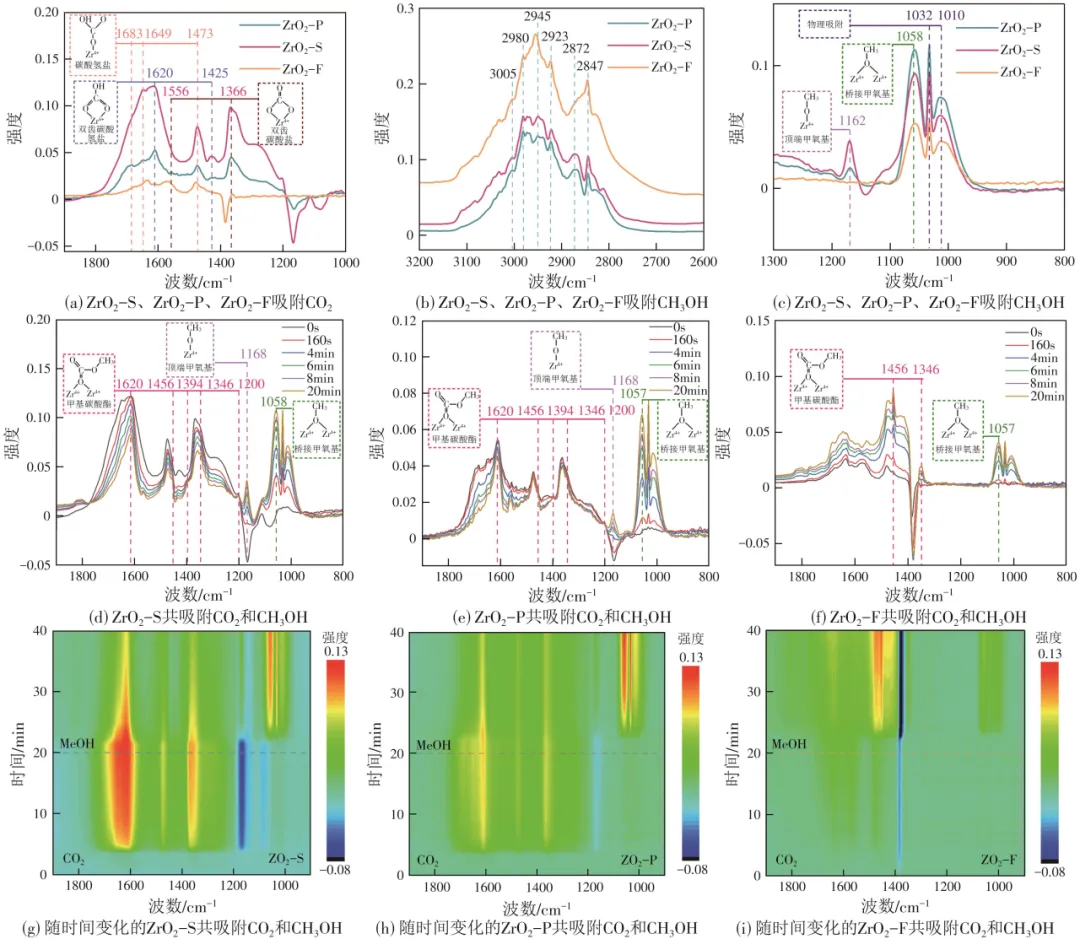
本文进一步采用原位FTIR来表征吸附物质的结构和活性位点的性质。通过DRIFTS揭示了不同形貌ZrO2上DMC合成的反应机理。图4(a)展示了以CO2为探针检测不同样品时的红外光谱。一般情况下,CO2与表面的—OH反应,以碳酸氢盐的形式吸附(1683cm-1、1649cm-1、1620cm-1、1473cm-1和1425cm-1),CO2还可以与表面晶格氧反应生成双齿碳酸盐(1556cm-1和1366cm-1)。由于不同形貌ZrO2表面的原子排列不同,因此当CO2传导到不同ZrO2纳米材料时,同一类型的碳酸盐物质也会表现出略有不同的振动频率。所以3种样品上均可形成双齿碳酸盐和碳酸氢盐物质。ZrO2-S上形成更多有利于反应的双齿碳酸盐物质,与其较高的OV浓度有关。3种样品上甲醇吸附物种的分布和分配如图4(b)、(c)所示。在图4(b)中,位于2945cm-1和2847cm-1处的峰分别归属于吸附在甲醇分子中CH3的弯曲振动和对称伸缩振动峰。在3005cm-1、2980cm-1和2872cm-1处的吸收峰对应CH3的不对称伸缩振动和对称伸缩振动峰,其根源是t-ZrO2上布朗斯特酸位点吸附的CH3OH被活化解离出*CH3。2923cm-1处的峰归属于单配位的甲氧基物种中的*CH3不对称伸缩振动,证实甲醇中的O—H键发生断裂解离出了甲氧基物种。在图4(c)中,1162cm-1和1058cm-1处的谱带分别为单齿和双齿甲氧基的峰,此外,1032cm-1和1010cm-1处的谱带可归属于甲醇的物理吸附。
图4(d)~(i)显示了当ZrO2预吸附CO2暴露于甲醇时的原位红外演变。随着甲醇吸附量的增加,出现新的峰(1620cm-1、1456cm-1、1394cm-1、1346cm-1和1200cm-1),可归属于甲基碳酸酯的峰,证实了甲醇吸附在催化剂上并参与反应。碳酸氢盐和双齿碳酸盐的峰强度减弱或者消失,也可证明甲醇与CO2参与了催化反应。同时,催化剂表面—OH的减少,证实了甲醇与催化剂表面—OH发生反应,生成了甲基碳酸酯中间体。有趣的是,随着甲醇不断通入,ZrO2-S和ZrO2-P中单齿和双齿甲氧基的峰在不断升高,ZrO2-F在1162cm-1处未能检测到能够与关键中间体反应的单齿甲氧基的弯曲振动峰,证实了ZrO2-F催化剂对CH3OH的活化能力较弱,可能与其较多的强酸性位点有关。较多的强酸性位点无法有效活化CO2和CH3OH为双齿碳酸盐和单齿甲氧基中间体,是其反应活性较低的主要原因。暴露稳定的(101)晶面和较多的弱酸碱位点和中强酸碱位点,可有效活化CO2与甲醇生成DMC的反应。
图4 443K下ZrO2上吸附CO2/CH3OH原位红外光谱
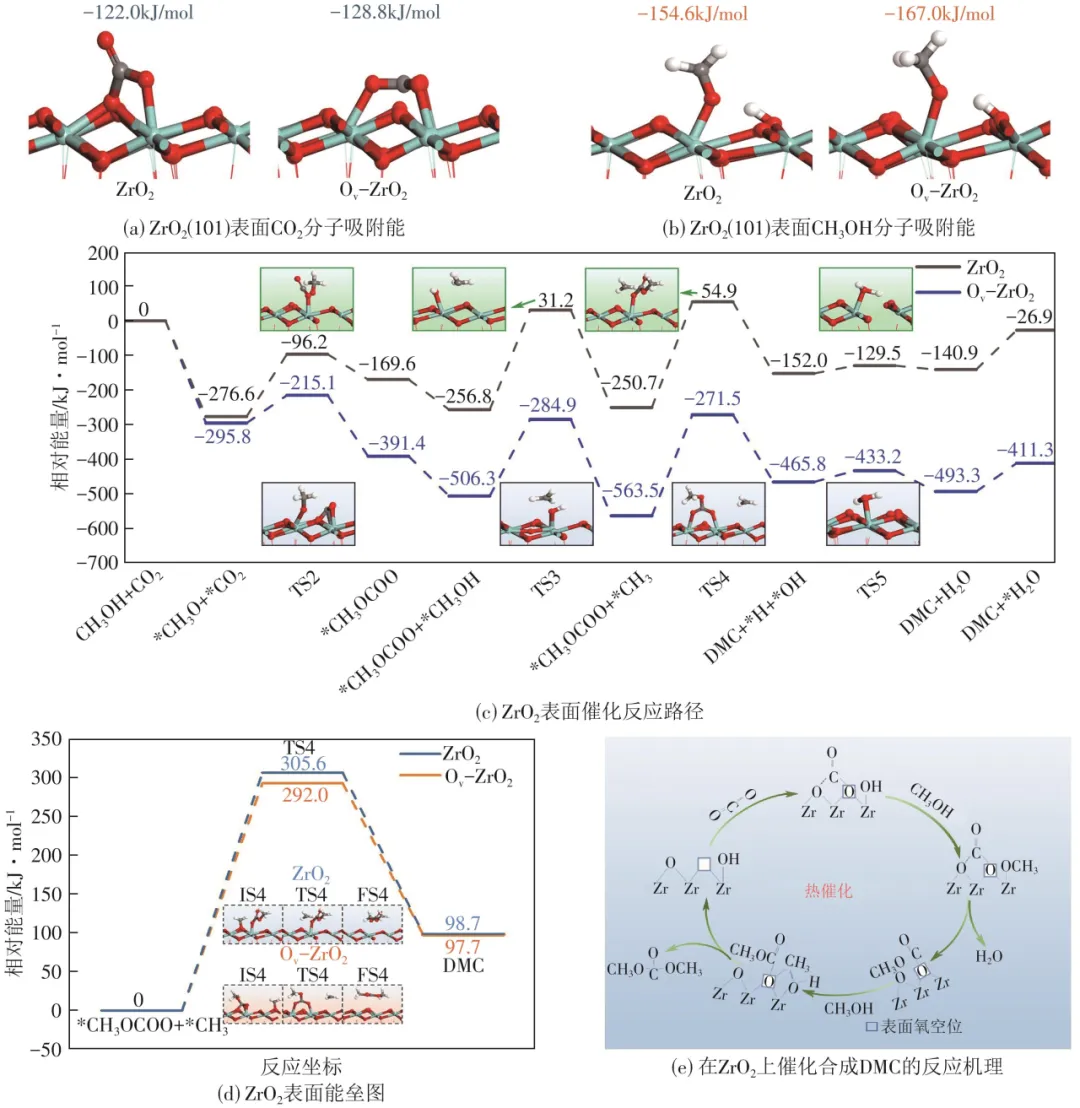
Ov-ZrO2(101)表面CO2分子吸附能较完美表面显著降低[图5(a)],表明OV可有效增强CO2的稳定吸附。当CH3OH吸附于ZrO2表面Zr1位点时,其O—H键发生自发解离生成*CH3O与*H物种,且该过程在Ov-ZrO2表面表现出更强的放热效应(ΔH=-167.0kJ/mol,完美表面ΔH=154.6kJ/mol)。结合原位红外光谱分析,提出ZrO2表面催化反应路径[图5(c)]:①表面Zr1位点吸附的CH3OH自发解离生成*CH3O;②*CH3O向*CO2靠近,生成以O端吸附于表面Zr1位点的关键中间体*CH3OCOO;③另一分子甲醇通过C—O键断裂形成以C端吸附于O2位点的*CH3;④*CH3向*CH3OCOO迁移生成目标产物DMC;⑤反应过程中产生的*H与*OH结合生成H2O,从而进行下一次循环反应。值得注意的是,引入OV显著降低了速率控制步骤(R4:*CH3OCOO+*CH3→DMC)的活化能垒,由完美表面的305.6kJ/mol降至292.0kJ/mol[图5(d)],降幅达13.6kJ/mol。基于以上表征结果和理论分析,本文提出了可行的ZrO2催化CO2/CH3OH直接合成DMC的反应机理[图5(e)]。首先,CO2的O原子在路易斯酸碱对相互作用下吸附在催化剂表面OV上,形成双齿碳酸盐物种。CO2分子中氧原子更容易在OV位置得电子从而吸附和活化。然后,为甲氧基物种的生成反应,Zr原子吸附的甲醇分子与OV附近的线性羟基作用形成甲氧基物种和一分子的水。之后,甲氧基物种与附近的双齿碳酸盐物种反应生成甲基碳酸盐中间体,而另一分子甲醇与氧空位附近的表面Zr反应形成甲基物种。最后,甲基物种与甲基碳酸盐中间体反应生成DMC,并在催化剂表面释放OV。
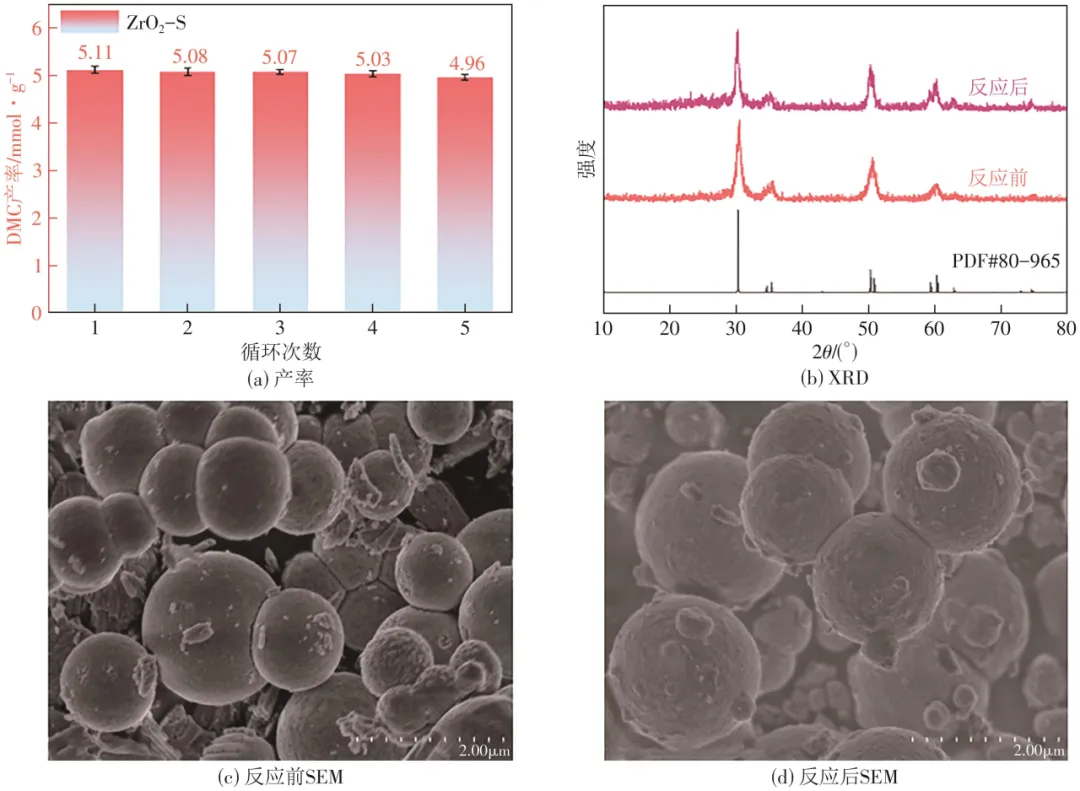
催化剂ZrO2-S经过5个反应周期后,DMC产率无明显变化,证明了催化剂性能的稳定性[图6(a)]。5次循环前后XRD图[图6(b)]和SEM图[图6(c)、(d)]进行对比,ZrO2-S的峰位置和峰强度以及球形结构表面均无明显变化,证实了催化剂结构的稳定性。
图6 5次循环反应前后的DMC产率、XRD和 SEM
通过SDS表面活性剂改性的球形ZrO2催化剂在CO2和甲醇合成DMC反应中表现出优异的催化性能。在最佳反应条件(反应温度170℃、CO2初始压力1.5MPa,催化剂用量0.05g,反应时间3h,不使用脱水剂)下,ZrO2-S催化剂催化CO2和甲醇合成DMC产率最高(5.11mmol/g)。经过5次循环实验,仍能保持较高的催化活性和结构形貌,证实了ZrO2-S催化剂的结构和性能稳定性。ZrO2-S较高的催化活性归因于高度暴露的(101)活性晶面以及较多的弱/中强酸碱位点以及较高的氧空位含量,促进了CO2和甲醇的活化生成关键中间体甲基碳酸酯。结合原位红外和理论计算研究,提出了ZrO2催化CO2和甲醇直接合成DMC的反应机理。研究结果可为CO2资源化利用直接合成DMC提供理论基础和科学依据。
第一作者:刘乐,硕士研究生,研究方向为二氧化碳转化与利用。
通信作者:张小超,教授,博士生导师,研究方向为二氧化碳转化与利用;张国强,副教授,研究方向为合成气及甲醇转化、生物质高值化利用。
邮发代号:82-311
订阅热线:010-64519502
网址:http://www.hgjz.com.cn