分子氧(O₂)活化生成活性氧物种(ROS)是催化氧化偶联过程的基础,然而传统方法依赖外部能量输入,这阻碍了可持续发展。
2026年05月11日,太原理工大学淡猛团队在Angewandte Chemie International Edition期刊发表题为“High-Entropy Catalyst Activated Molecular Oxygen for Oxidative Coupling Under Ambient Conditions”的研究论文,团队成员齐润龙为论文第一作者,淡猛、广东第二师范学院郭臻为论文共同通讯作者。
第一作者:齐润龙
通讯作者:淡猛、郭臻
通讯单位:太原理工大学、广东第二师范学院
论文DOI:10.1002/anie.1664539
该研究首次证明,具有丰富自生晶格畸变的多元素协同非贵金属高熵硫化物纳米晶(HESNCs)能够在环境条件下自发活化分子氧(O₂)生成超氧自由基(·O₂⁻),并驱动多种氧化偶联反应(S-N、C-S、C-N),具有高催化活性和卓越稳定性(>300天)。同时,HESNCs驱动的无外场催化方案可实现优异产率(>90%)的药物合成,并可放大至摩尔级生产,展现出巨大的工业应用潜力。实验和计算分析揭示,HESNCs中严重的本征晶格畸变调控了d带中心并增强了泡利排斥,使得电子从有机供体(如胺)转移到O₂,生成·O₂⁻和供体自由基(Don·)。随后,·O₂⁻诱导底物氧化形成偶联活性中间体(Sub·),这些中间体与Don·结合生成氧化偶联产物,同时将·O₂⁻还原为水。该研究建立了一种新型“电子供体辅助高熵催化剂(HECs)介导”的环境条件O₂活化范式,无需外部能量输入即可实现氧化偶联反应。
氧化偶联反应作为有机合成中的基本方法学,通过外部氧化剂介导的电子转移连接亲核中心,用于生产重要的工业产品和药物。空气中的分子氧(O₂)作为氧化偶联反应的理想氧化剂,具有天然丰富、成本低廉和环境兼容等无与伦比的优势。然而,在环境条件下实际应用O₂主要受到自旋禁阻反应路径的限制。活化O₂生成活性氧物种(ROS,例如单线态氧(¹O₂)、超氧阴离子(·O₂⁻)、羟基自由基(·OH))为克服O₂的固有弱点并引发氧化偶联提供了一种有效策略。在这些ROS中,·O₂⁻由于具有合适的氧化还原电位(2.4V vs. RHE)而表现出 卓越选择性,显著降低了不希望的过氧化风险。此外,其延长的半衰期(51-422秒)和宽pH耐受范围(pH=2-10)确保了在不同反应环境中的可靠催化性能,巩固了其作为受控氧化偶联反应最佳ROS的地位。
直接将O₂活化为·O₂⁻对于实现环境友好且廉价的环境条件氧化偶联反应过程极具吸引力。然而,O₂的高活化能垒导致其产生¹O₂高度依赖于外部能量输入(例如光、热或电能),即使存在催化剂也是如此,导致操作复杂、效率低且能耗高,很大程度上阻碍了工业放大。理论上,直接将O₂催化活化为·O₂⁻几乎是不可能的,因为所需的显著电子转移通常会导致催化剂氧化,引起不可逆失活。因此,在不需外部能量输入的情况下直接将O₂活化为·O₂⁻以引发环境条件氧化偶联反应仍然是一项艰巨的挑战。
芬顿反应能够在环境条件下产生ROS,通过利用电子供体有效构建完整的循环路径,防止催化位点失活。在氧化偶联反应中,众多底物都显示出作为有效电子供体的潜力。受此启发,该研究报道了可持续催化领域的一项突破:具有丰富本征晶格畸变的多元素协同非贵金属基高熵硫化物纳米晶(HESNCs: FeCoCuZnCdS),能够在无需外部能量输入的情况下实现自发的O₂活化和底物氧化偶联。HESNCs在环境条件下,对于多种底物(超过85个实例)形成S-N、C-S和C-N键展现出卓越的催化活性和稳定性(>300天),同时保持了优异的可放大性至摩尔级合成,突显了其巨大的工业转化潜力。实验和计算分析揭示,具有独特多元素协同作用的HESNCs介导电子从电子供体(底物)转移到分子氧(O₂),从而在环境条件下生成·O₂⁻并驱动后续的氧化偶联反应。
图1 策略发展。已开发的用于O₂活化的策略以及本研究中描述的方法,在该方法中O₂在环境条件下被活化为∙O₂⁻用于各种氧化偶联反应。
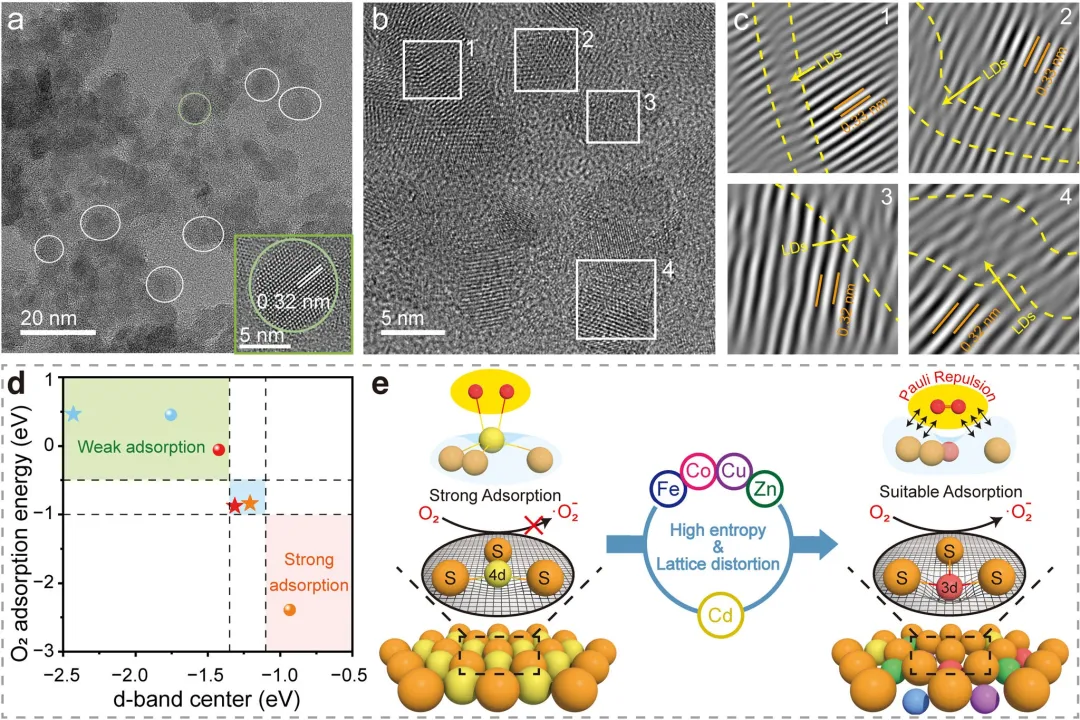
图2 FeCoCuZnCdS高熵硫化物纳米晶(HESNCs)的结构特征及DFT计算。(a)HESNCs的TEM图像。(b)高分辨TEM结果及(c)对应区域的IFFT图。DFT计算:(d)O₂在MSs(球)和HESNCs(五角星)上的吸附能。Cu:蓝色; Co:红色; Fe:橙色。(e)HESNCs中的晶格畸变和泡利排斥削弱了O₂吸附。4d金属:黄色; S:橙色; 3d金属:蓝色、紫色、浅红色和绿色; O:红色。
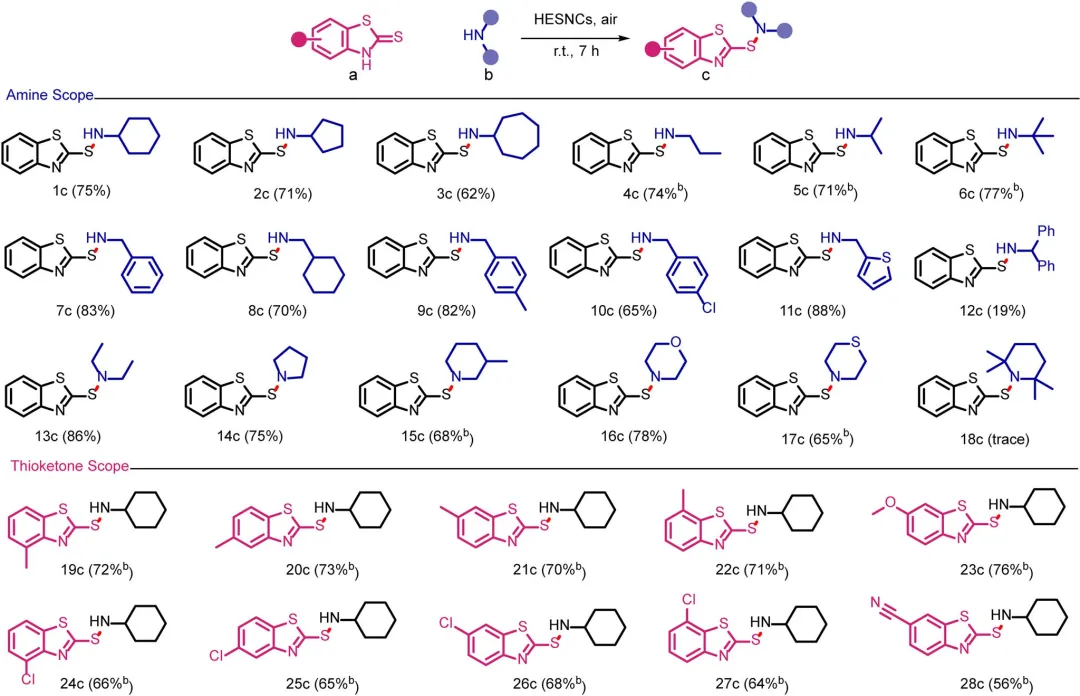
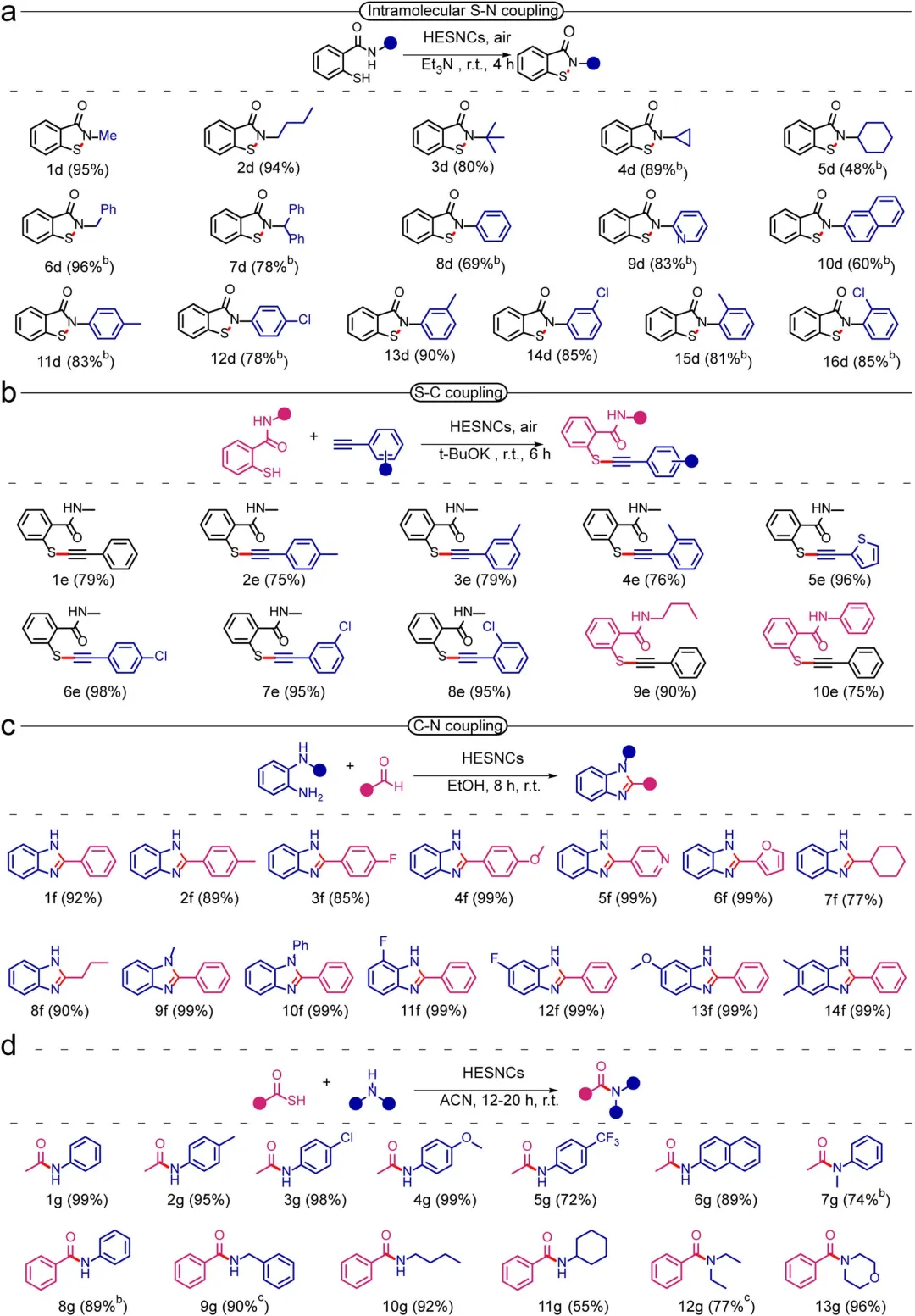
图3 HESNCs催化的S─N氧化偶联的底物范围。反应条件:硫酮(0.1mmol)、胺(0.5mmol)、HESNCs(5mg)、2mL乙腈、室温、空气氛围、7h。b 1mmol胺。所有产率均为分离产率。
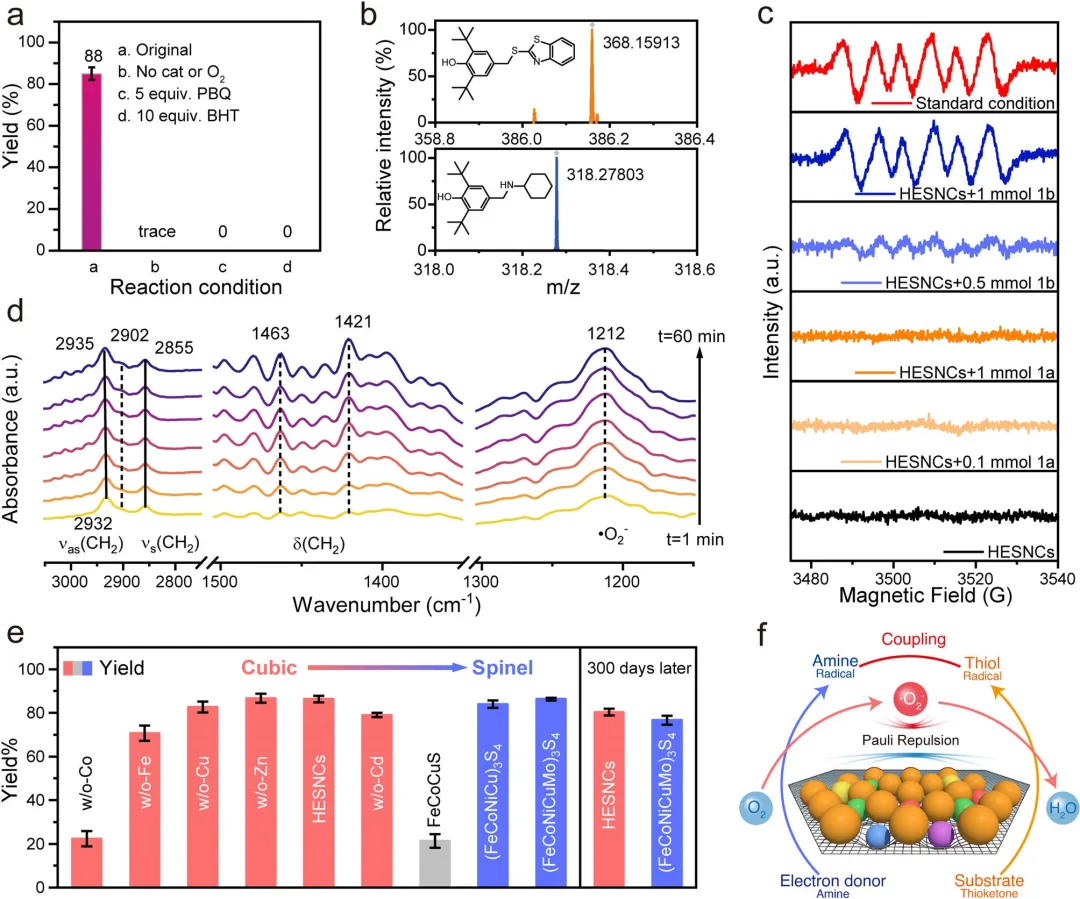
图4 反应机理研究。(a)对照实验。(b)用BHT进行的捕获实验。(c)不同条件下的原位EPR谱图。(d)操作条件下HESNCs上S─N氧化偶联的原位FT-IR谱图(0.1mmol硫酮、0.5mmol胺、5mg HESNCs、2mL乙腈、室温)。(e)各种金属硫化物的S─N氧化偶联性能。(f)无需外部能量输入的“胺辅助HESNCs介导”环境条件O₂活化用于S─N氧化偶联反应的机理。产率通过HPLC以联苯为内标测定。
图5 HESNCs催化的S─N、C─S和C─N键形成中获得的产物产率。(a)分子内S─N形成的范围。(b)C─S形成的范围。(c)苯并咪唑(C─N形成)的范围。(d)酰胺(C─N形成)的范围。(a)的反应条件:标准条件为HESNCs(5mg)、2-巯基苯甲酰胺底物(0.1mmol)、Et₃N(0.2mmol)、2mL EtOH、室温、空气氛围、4h。b 2mL N,N-二甲基甲酰胺(DMF)。(b)的反应条件:HESNCs(5mg)、2-巯基苯甲酰胺(0.1mmol)、苯乙炔(0.1mmol)、tBuOK(0.25mmol)、2mL EtOH、室温、空气氛围、6h。(c)的反应条件:5mg HESNCs、醛(0.1mmol)、邻苯二胺(0.1mmol)、2mL EtOH、室温、空气氛围、8h。(d)的反应条件:标准条件为硫代酸底物(0.2mmol)、胺(0.1mmol)、2mL乙腈、室温、空气氛围、12h。b 20h。c 硫代酸底物(0.1mmol)、胺(0.4mmol)。所有产率均为分离产率。
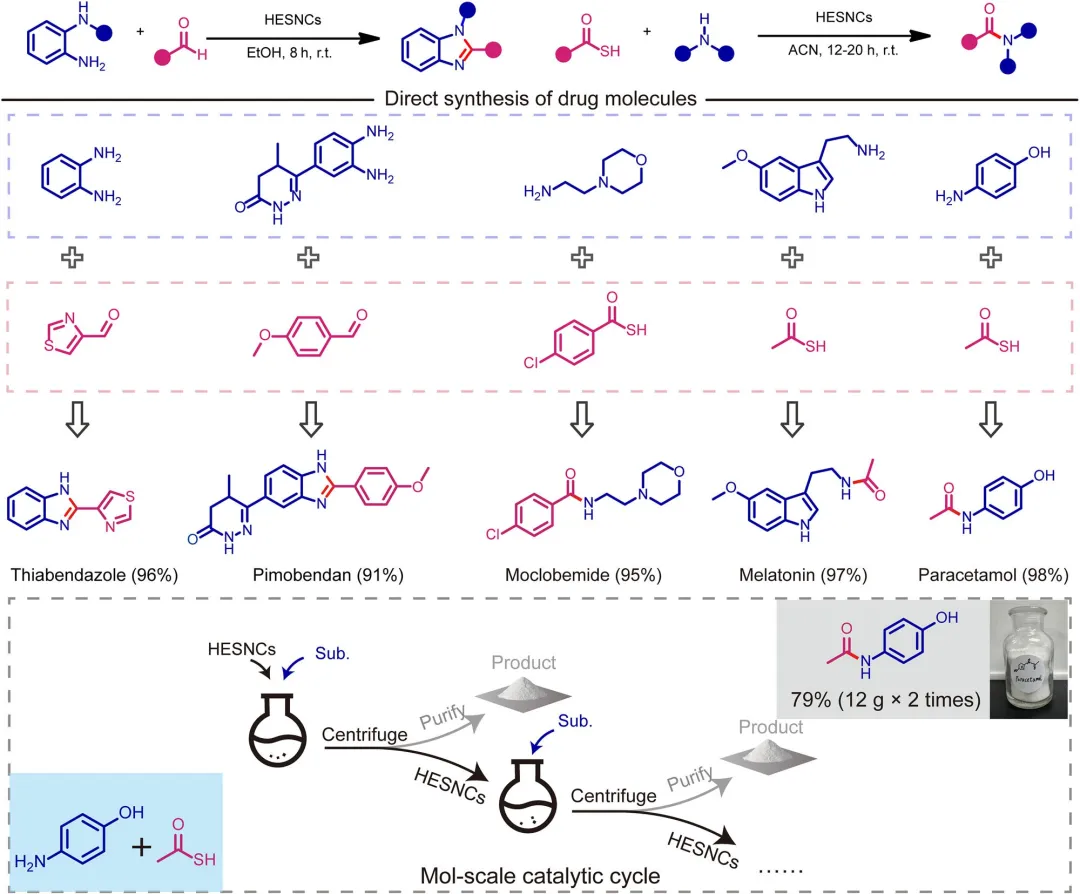
图6 药物分子的直接合成及摩尔级实验。反应条件1(苯并咪唑的合成):HESNCs(5mg)、苯甲醛(0.1mmol)、邻苯二胺(0.1mmol)、2mL EtOH、室温、空气氛围、8h。反应条件2(酰胺的合成):5mg HESNCs、硫代酸底物(0.2mmol)、胺(0.1mmol)、2mL乙腈、室温、空气氛围、12h。对于摩尔级实验:1g HESNCs、硫代酸底物(0.2mol)、胺(0.1mol)、200mL乙腈,在室温和空气氛围下反应60h。所有产率均为分离产率。
该研究开发了非贵金属HESNCs,其具有自生晶格畸变和优化的d带中心位置,能够自发活化O₂并在无需外部能量输入的环境条件下驱动一系列氧化偶联反应。HESNCs对S-N键、C-S键和C-N键的形成显示出卓越的催化活性(产率高达99%)、宽温度适应性(0°C以上产率无明显下降)、出色的可回收性(多次循环后产率从88%降至83%),并在药物合成中展示了可放大性(24.1g,79%产率)。
总之,该研究提出了一种通用的“电子供体辅助HECs介导”的O₂活化新范式,用于在环境条件下进行多种氧化偶联反应(S-N、C-S、C-N等)。这种无外场催化方案展示了其应用潜力。未来研究应优先考虑:(i) 放大反应并拓展底物范围;(ii) 开发HECs以选择性生成其他ROS(¹O₂、·OH等),从而在环境条件下实现更多样的氧化偶联反应。


 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
