太原科技大学/太原理工大学刘一鸣教授团队:原子级晶格匹配 S 型异质结实现甘油高效高选择性光电催化转化
- 2026-03-26 17:46:31

DOI:10.1016/S1872-2067(26)64955-8


近日,《催化学报》在线发表了太原科技大学/太原理工大学刘一鸣教授团队,太原理工大学王剑副教授在光电催化生物质高值化利用领域的最新研究成果。该工作通过晶格匹配工程策略,构建了具有原子级连贯界面的h-WO3/TiO2 S型异质结光阳极,实现了生物柴油副产物甘油向高附加值二羟基丙酮(DHA)的高效、高选择性光电催化转化。
论文第一作者为太原理工大学张王刚副教授,论文共同通讯作者为太原理工大学王剑副教授、太原科技大学/太原理工大学刘一鸣教授。


随着全球能源消费持续增长,开发绿色可持续的能源转化技术成为研究核心热点。光电催化(PEC)技术可直接利用丰富的太阳能实现光能到化学能的转化,是生物质资源高值化利用的重要前沿路径。生物柴油产业每年产生巨量副产物甘油,将其定向转化为高附加值的二羟基丙酮(DHA),兼具固碳环保效益与产业经济价值,是生物质资源化利用的重要方向。
然而,光电催化甘油转化过程长期面临两大核心瓶颈:一是传统TiO2基光阳极材料光生载流子复合严重,光 - 化学能转化效率低;二是甘油氧化反应路径复杂,易发生 C-C 键断裂和过度氧化,目标产物 DHA 选择性差。构建 WO3/TiO2异质结是解决上述问题的有效策略,但现有研究多聚焦于宏观结构调控,对异质结原子级界面晶格匹配的关键作用,及其对界面内建电场、电荷转移行为的影响机制尚未被系统阐明,晶格失配引发的界面缺陷、弱内建电场等问题,始终制约着异质结电荷分离效率与催化选择性的同步提升。


1. 开发退火调控的晶格匹配工程策略,在金红石TiO2纳米棒上实现六方相WO3的近外延生长,构建了原子级连贯界面的h-WO3/TiO2异质结,晶格失配率低至0.027%,远低于单斜相WO3/TiO2体系的 2.30%。
2. 揭示了原子级晶格匹配对异质结内建电场的强化机制,构建的S型异质结实现90%的光生载流子复合抑制,大幅优化了光电催化反应动力学。
3. 阐明了晶格匹配界面对甘油仲羟基的选择性吸附调控机制,打破传统甘油氧化的选择性瓶颈,DHA选择性达35%,是单斜相异质结体系的2.19倍,同时实现甘油转化率较单斜相体系提升40%以上。


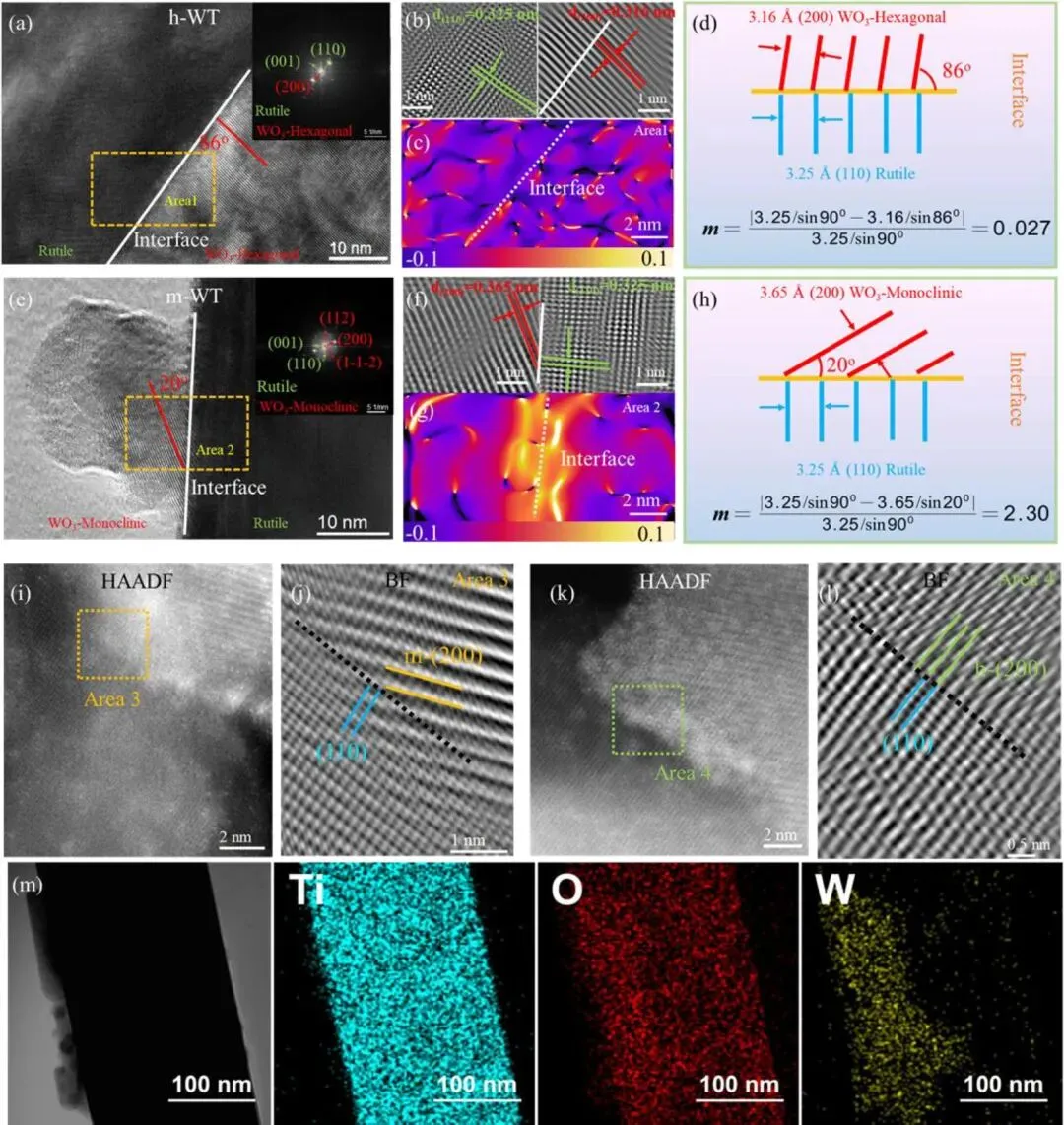
图1. (a) h-WT 的高分辨透射电子显微镜(HRTEM)图像;(b) 区域 1 的界面放大图像。(a) 中的插图为区域 1 的快速傅里叶变换(FFT)花样。(c) 区域 1 沿 eₓ矢量方向的几何相位分析(GPA)结果。(d) 用于计算 TiO₂与 h-WO₃之间晶格失配率(m)的示意图。(e) m-WT 的 HRTEM 图像;(f) 区域 2 的界面放大图像。(f) 中的插图为区域 2 的 FFT 花样。(g) 区域 2 沿 eₓ矢量方向的 GPA 结果。(h) 用于计算 TiO₂与 m-WO₃之间晶格失配率(m)的示意图。(i)(j) 区域 3 方框区域的放大球差校正高角环形暗场(AC-HAADF)图像与球差校正明场(AC-BF)图像。(k)(l) 区域 4 方框区域的放大 AC-HAADF 图像与 AC-BF 图像。(m) h-WT的扫描透射电子显微镜-能谱(STEM-EDS)元素面分布结果。
要点:
1. 高分辨透射电子显微镜(HRTEM)表征证实,六方相 WO3/TiO2异质结(h-WT)形成了原子级清晰、连续的连贯界面,而单斜相体系(m-WT)界面存在明显的晶格无序与结构缺陷。
2. 几何相位分析(GPA)与定量计算显示,h-WT 异质结晶格失配率仅为 0.027%,实现了六方相 WO3在 TiO2纳米棒上的近外延生长;而 m-WT 体系晶格失配率高达 2.30%,界面存在显著的晶格应变积累。
3. 密度泛函理论(DFT)计算表明,h-WT 异质结界面形成能低至 - 3.22 eV,远低于 m-WT 体系的 - 2.23 eV,证实晶格匹配界面具有更优异的结构稳定性与界面电子耦合作用。
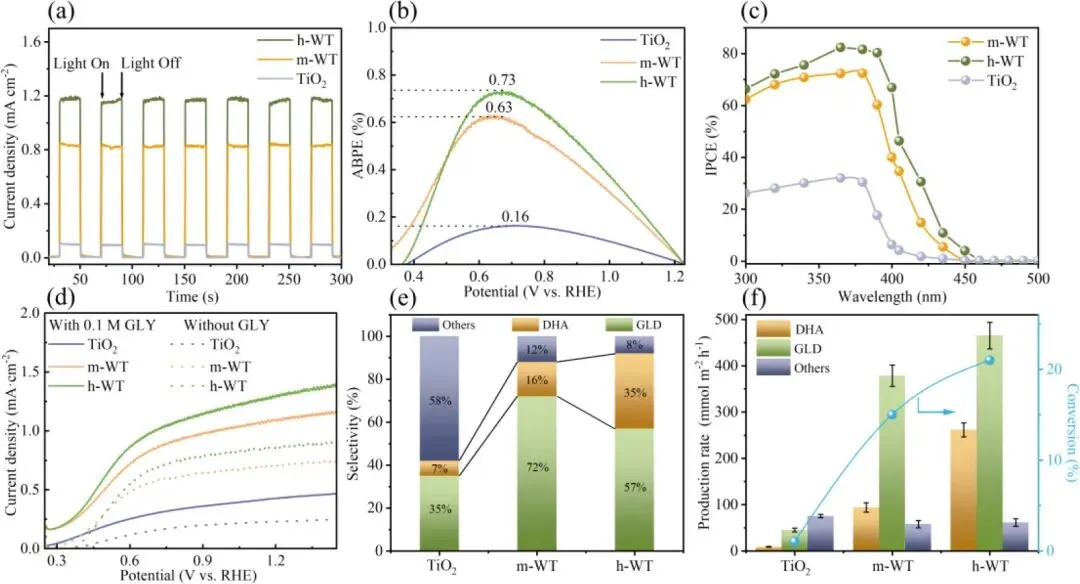
图2. (a) 在 AM 1.5G 模拟太阳光照射下,TiO₂、m-WT 和 h-WT 样品在含 0.1 mol・L⁻¹ 甘油(Gly)的 0.1 mol・L⁻¹ Na₂SO₄电解液中,1.23 V vs. RHE 电位下的瞬时光电流密度 - 时间曲线;(b) 各样品的光电转换效率(ABPE)数值;(c) 各样品的入射光子-电流转换效率(IPCE)光谱。(d) TiO₂、m-WT 和 h-WT 光阳极的线性扫描伏安(LSV)曲线。(e) 1.23 V vs. RHE 电位下,TiO₂、m-WT 和 h-WT 光阳极上有机产物的选择性。(f) TiO₂、m-WT 和 h-WT 光阳极上 DHA 与 GLD 的生成速率。
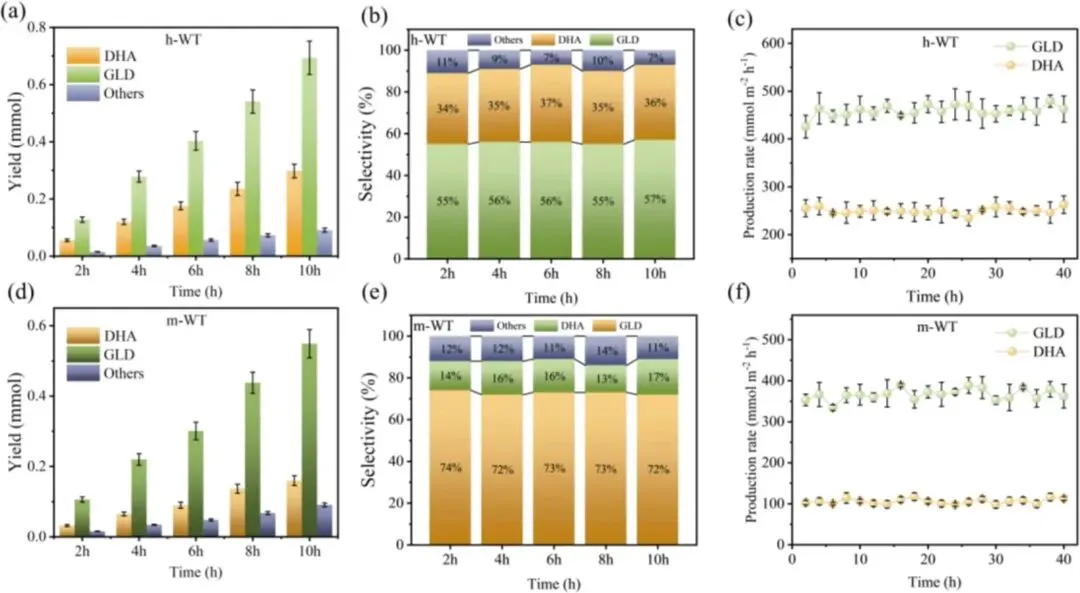
图3. 1.23 V(vs. RHE)条件下,h-WT (a) 与 m-WT (d) 光电阳极上光电化学甘油氧化反应(PEC GOR)的动力学曲线。1.23 V(vs. RHE)条件下,h-WT (b) 与 m-WT (e) 光电阳极上有机产物的选择性(测试时长为 10 h)。h-WT (c) 与 m-WT (f) 光电阳极在光电化学甘油氧化反应(PEC GOR)中的稳定性。(误差线代表三次平行重复实验结果的标准偏差,偏差值在 10% 以内;图中对应数据为平均值)
要点:
1. 光电性能测试显示,在 1.23 V vs. RHE 电位下,h-WT 光阳极的光电流密度达 1.3 mA cm⁻²;其光电转换效率(ABPE)与入射光子-电流转换效率(IPCE)分别达 0.73% 和 82%,较 m-WT 与纯 TiO2实现显著提升。
2. 高效液相色谱(HPLC)分析结果表明,h-WT 光阳极的 DHA 选择性达 35%,同时其总 C3 产物选择性超 90%,有效抑制了甘油 C-C 键断裂与过度氧化副反应。
3. 稳定性测试结果显示,异质结光阳极在连续 40 h 的反应中,产物生成速率无明显衰减,反应后形貌与晶相结构保持稳定,具备优异的长期运行稳定性。
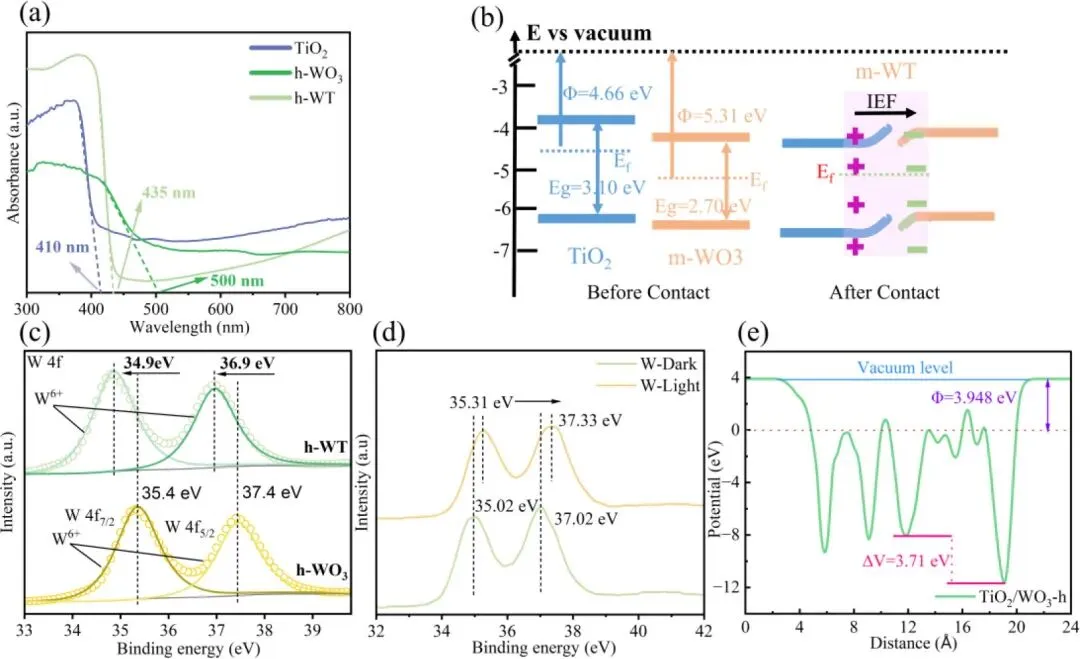
图4. (a) TiO₂、m-WO₃与 h-WO₃的紫外 - 可见漫反射光谱(UV‑vis DRS)。(b) h-WO₃与 TiO₂的能级结构图。(c) TiO₂与 h-WT 的高分辨 Ti 2p X 射线光电子能谱(XPS)。(d) h-WO₃与 h-WT 的高分辨 W 4f X 射线光电子能谱(XPS)。(e) h-WT 的电势计算。
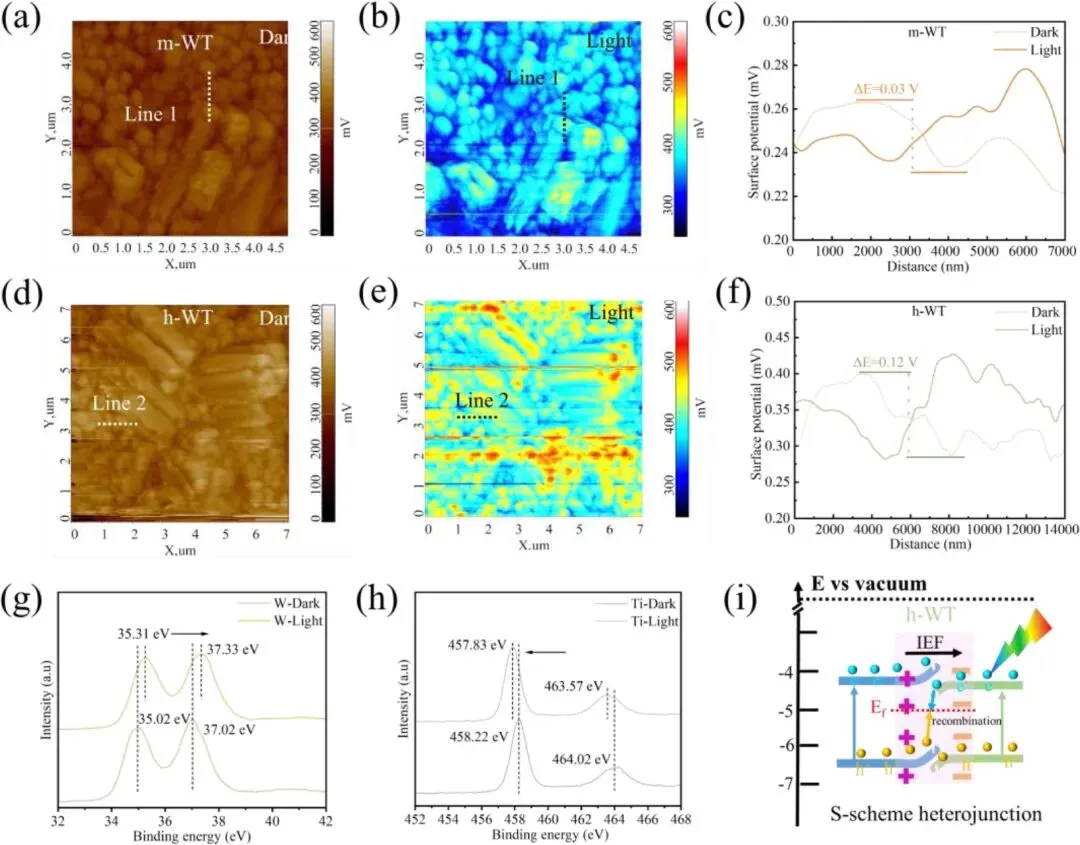
图5. (a,b) 分别为黑暗与光照(AM 1.5G,100 mW cm⁻²)条件下 m-WT 的开尔文探针力显微镜(KPFM)表面电势图。(c) 黑暗与光照条件下,沿 m-WT 界面的表面电势线扫图谱(对应图 a、b 中的曲线 1)。(d,e) 分别为黑暗与光照条件下 h-WT 的开尔文探针力显微镜(KPFM)表面电势图。(f) 黑暗与光照条件下,沿 h-WT 界面的表面电势线扫图谱(对应图 d、e 中的曲线 2)。(g,h) 黑暗与光照条件下 h-WT 的 W 4f (g) 与 Ti 2p (h) 高分辨 X 射线光电子能谱(XPS)。(i) 光照条件下 h-WO₃与 TiO₂之间 S 型电荷转移机理的示意图。
要点:
1. 紫外光电子能谱(UPS)与 X 射线光电子能谱(XPS)表征证实,h-WT 异质结的界面内建电势高达 3.71 eV,为界面电荷转移提供了强驱动力。
2. 原位光照 XPS 与开尔文探针力显微镜(KPFM)结果直接验证了异质结的 S 型电荷转移路径:光生电子在TiO2导带富集,光生空穴在 WO3价带富集,在实现高效载流子空间分离的同时,保留了材料的强氧化还原能力。
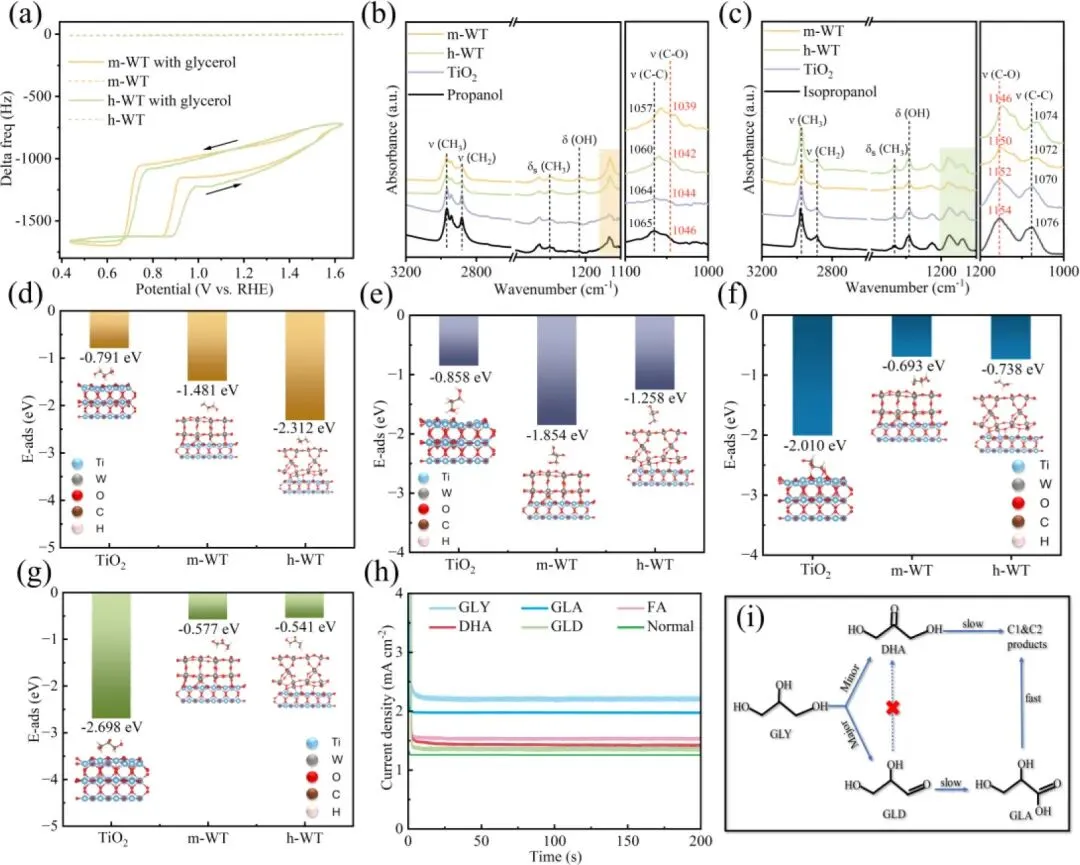
图6. (a) TiO₂、m-WT 与 h-WT 光电阳极吸附异丙醇和正丙醇后的电化学石英晶体微天平(EQCM)曲线;(b、c) 上述对应样品的傅里叶变换红外光谱(FTIR)。(d、e) 基于密度泛函理论(DFT)计算得到的甘油(Gly)通过伯羟基或仲羟基吸附于 TiO₂、m-WT 和 h-WT 表面的吸附能。(f、g) 甘油醛(GLD)与二羟基丙酮(DHA)在 TiO₂、m-WT 和 h-WT 表面的吸附能。(h) AM 1.5G 模拟太阳光照射下,在含 0.1 mol・L⁻¹ 甘油(GLY)、甘油醛(GLD)、甘油酸(GLA)或二羟基丙酮(DHA)的 0.1 mol・L⁻¹ 硫酸钠(Na₂SO₄)电解液中,h-WT 光电阳极在 1.23 V(vs. RHE,相对于可逆氢电极)下的电流密度 - 时间曲线。(i) 本文提出的甘油氧化反应(GOR)的反应路径。
要点:
1. 电化学石英晶体微天平(EQCM)测试表明,h-WT 与 m-WT 对甘油的总吸附量相近,证明二者产物选择性差异并非源于甘油总吸附能力,而是对甘油不同羟基的特异性吸附作用。
2. 以正丙醇、异丙醇为分子探针的傅里叶变换红外光谱(FTIR)结果显示,h-WT 对模拟仲羟基的异丙醇 ν(C-O) 振动峰红移达 8 cm⁻¹,远大于 m-WT 的 4 cm⁻¹,证实 h-WT 对甘油仲羟基的吸附亲和力显著更强;而 m-WT 对伯羟基的吸附作用更突出,产物选择性更偏向 GLD。
3. DFT 计算进一步量化了吸附能差异,h-WT 对甘油仲羟基的吸附能低至 - 1.854 eV,显著高于 m-WT(-1.258 eV)与纯TiO2(-0.858 eV);同时 DHA 与 GLD 在 h-WT 表面吸附能极弱,可快速脱附,有效避免了产物过度氧化,最终实现超 85% 的总 C3 产物选择性。
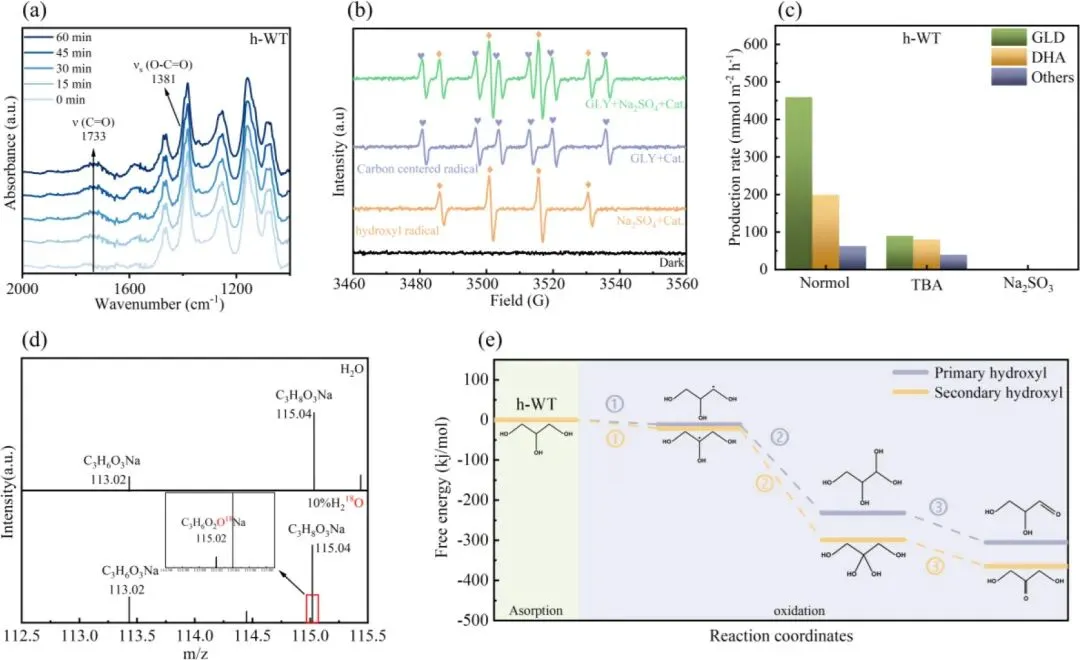
图7. (a) TiO₂、(b) h-WT 在 AM 1.5G(100 mW・cm⁻²)模拟太阳光照射 60 min 过程中,异丙醇动态氧化反应的傅里叶变换红外光谱(FTIR)。(c) 室温条件下,h-WT 在不同电解液中的电子自旋共振谱(ESR)。(d) 光电化学甘油氧化反应(PEC GOR)在含 10% H₂¹⁸O 的同位素标记电解液中,生成的二羟基丙酮(DHA)产物的液相色谱 - 质谱联用谱(LC-MS)。(e) h-WT 表面涉及伯羟基与仲羟基的氧化过程的吉布斯自由能变化曲线。
要点:
1.原位光照 FTIR 光谱显示,h-WT 催化异丙醇氧化过程中,仅出现丙酮的 C=O 特征峰,未检测到代表 C-C 键断裂的羧酸盐产物信号,直接证实晶格匹配的异质结可完全抑制甘油氧化过程中的 C-C 键断裂,实现高选择性的羰基化反应。
2.电子顺磁共振(ESR)测试与自由基捕获实验证实,h-WT 体系的羟基自由基(・OH)信号强度显著高于 m-WT 与纯TiO2,且加入・OH 捕获剂后,DHA 与 GLD 的生成率显著下降,证明催化剂表面吸附的・OH 是甘油选择性氧化的核心活性物种,而非光生空穴直接氧化路径。
3.H218O 同位素标记实验结果显示,生成的 DHA 分子中 C=O 基团的氧原子来自体系中的 H2O,证实表面吸附的・OH 通过偕二醇中间体路径参与甘油仲位 C-H 键的选择性断裂,最终经脱水反应生成 DHA,明确了甘油氧化的分子级反应路径。
4.DFT 吉布斯自由能计算揭示了 h-WT 高 DHA 选择性的热力学本质:甘油仲羟基氧化生成 DHA 的路径,最高反应能垒比伯羟基氧化生成 GLD 的路径低 66.8 kJ・mol⁻¹,证明仲羟基氧化生成 DHA 在热力学上更具优势。


研究团队通过多尺度原位表征与 DFT 理论计算,系统揭示了晶格匹配 S 型异质结催化甘油定向氧化的内在机制:
1. S 型电荷转移的高效载流子分离机制:原子级晶格匹配大幅强化了异质结界面内建电场,驱动光生载流子沿 S 型路径高效转移,显著抑制了光生载流子的无效复合,为甘油氧化反应提供了充足的光生空穴与活性自由基。
2. 仲羟基选择性吸附的反应路径调控机制:晶格匹配优化的界面电子结构,显著增强了对甘油仲羟基的吸附亲和力(吸附能达 - 1.854 eV),优先活化甘油仲位 C-H 键,从热力学上驱动反应向 DHA 生成的方向进行,实现了产物选择性的精准调控。
3. 自由基主导的选择性氧化机制:原位傅里叶变换红外光谱(in-situ FTIR)、电子顺磁共振(ESR)与同位素标记实验证实,甘油氧化反应主要由催化剂表面吸附的羟基自由基(・OH)主导,通过偕二醇中间体路径选择性断裂仲位 C-H 键生成 DHA,而非直接空穴氧化,有效避免了 C-C 键断裂与过度氧化副反应的发生。
1. 本工作开发了一种晶格匹配工程策略,通过退火温度精准调控,成功构建了具有原子级连贯界面的六方相 WO3/TiO2 S 型异质结,实现了 0.027% 的超低晶格失配,为异质结光阳极的界面工程设计提供了新思路。
2. 原子级晶格匹配可显著强化异质结界面内建电场,优化 S 型电荷转移路径,实现载流子分离效率与寿命的大幅提升,同时增强对甘油仲羟基的选择性吸附,同步解决了光电催化甘油转化中转化效率低与产物选择性差的两大核心难题。
3. 最优的 h-WT 光阳极实现了 35% 的 DHA 选择性、40% 的甘油转化率提升,以及超 40 h 的运行稳定性,其甘油转化速率达 788.6 mmol m⁻² h⁻¹,为生物质高值化利用的高效光阳极设计提供了重要的理论基础与实验支撑。


刘一鸣,男,工学博士,太原科技大学化学工程与技术学院教授、太原理工大学环境科学与工程学院教授,催化转化与能源耦合山西省重点实验室主任,本论文共同通讯作者。山西省高层次青年人才,山西省学术技术带头人,山西省三晋英才拔尖骨干人才,山西省优秀科技工作者。 省级重点实验室、工程研究中心主任。《Engineered Science》和《燃料化学学报》期刊青年编委。
王剑,男,中共党员,山西昔阳人,副教授,本论文共同通讯作者。现在太原理工大学材料科学与工程专业从事科研与教学工作。曾获山西省科技进步二等奖一项。主持国家自然科学青年项目、中国博士后基金面上资助、山西省重点研发计划、山西省自然科学基金、山西省科技合作交流专项及山西省综合教育改革项目等省部级以上项目多项,发表SCI期刊论文60余篇(第一/通讯30余篇),申请/授权国家发明专利10余项(第一发明人7项)。
张王刚,男,山西曲沃人,副教授,本论文第一作者。现于太原理工大学材料科学与工程学院从事教学与科研工作。 曾获山西省科技进步二等奖一项、山西省科学技术三等奖一项。 主持/参与多项国家及省部级项目,发表SCI期刊论文40余篇,申请/授权国家发明专利10余项。
文献信息:
Wanggang Zhang, Haochen Xie, Hongliang Wang, Rufeng Tian, Lei Liu, Jian Wang*, Yiming Liu*, Chin. J. Catal., 2026, 82, 161-173. (点击链接到Elsevier网站,下载全文)






长按二维码关注我们 ▶▶▶

Chinese Journal of Catalysis(《催化学报》,月刊,英文刊)创刊于1980年,是中国化学会催化专业委员会会刊,由中国科学院大连化学物理研究所和中国化学会共同主办。期刊现任共同主编为李灿院士和张涛院士,主要发表催化领域各研究方向的最新研究成果,SCI影响因子为17.7,入选中国科学院期刊分区化学类1区Top期刊,连续13年被评为“中国最具国际影响力学术期刊”,入选中国科技期刊卓越行动计划二期项目英文领军期刊,入选中国科学院精品科技期刊建设试点项目。期刊所有文章均不收取审稿费和版面费等任何费用,电子版在Elsevier平台出版:http://www.journals.elsevier.com/chinese-journal-of-catalysis。
如有任何疑问,请联系编辑部(cjcatal@dicp.ac.cn)


