图文导读
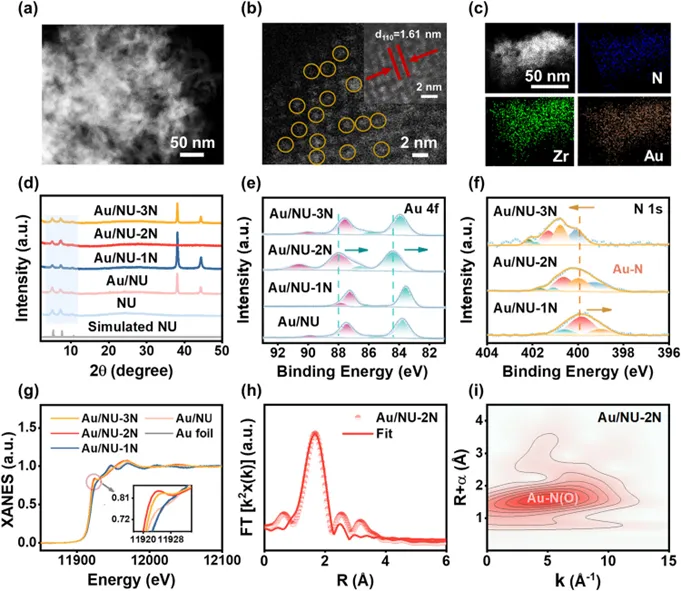
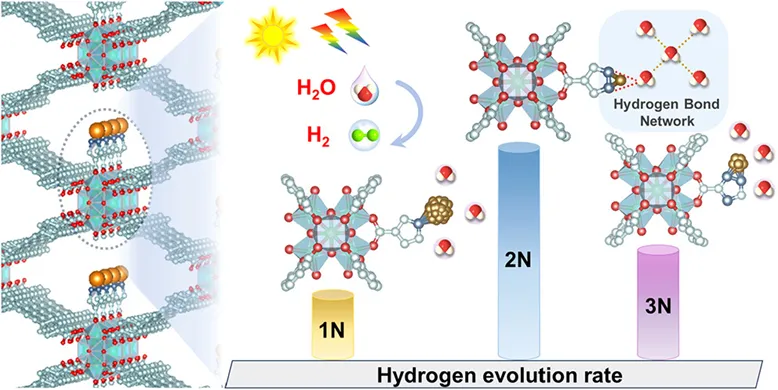
Fig. 2.(a, b) HAADF-STEM images (orange circles represent Au single atoms) and (c) elemental mappings of Au/NU-2N. (d) PXRD patterns of simulated NU, NU, Au/NU, Au/NU-1N, Au/NU-2N, and Au/NU-3N. (e) Au 4f XPS spectra of Au/NU, Au/NU-1N, Au/NU-2N, and Au/NU-3N. (f) N 1s XPS spectra of Au/NU-1N, Au/NU-2N, and Au/NU-3N. (g) Au L3-edge XANES spectra of Au foil, Au/NU, Au/NU-1N, Au/NU-2N, and Au/NU-3N. (h) Au L3-edge EXAFS spectra of Au/NU-2N and its corresponding fitting curve. (i) The Au L3-edge WT-EXAFS plots of Au/NU-2N.
选用锆基金属有机框架NU-901(简称NU)作为模型材料,其具有优异的光活性与稳定性。该结构由TBAPy配体与Zr-oxo团簇构成,每个团簇与八个TBAPy配体及四个调节剂配位,而每个TBAPy又连接四个Zr团簇。为构筑Au配位结构,分别合成了Au/NU、Au/NU-1N、Au/NU-2N和Au/NU-3N。首先通过溶剂热法合成NU,随后通过溶剂辅助配体引入(SALI)法将不同含氮杂环配体(1N、2N、3N)接枝到Zr–O团簇上,得到NU-1N、NU-2N和NU-3N。1H NMR结果表明,配体交换充分发生,且其比例可通过调控引入配体量实现精确控制。进一步负载Au后得到对应催化剂。ICP-OES测得Au含量分别为9.97、10.74和6.85 wt%,配体与Au的摩尔比分别约为2.67、2.45 和4.00,表明Au/NU-2N中存在未配位的2N单元。
扫描电子显微镜(SEM)分析表明,Au/NU、Au/NU-1N、Au/NU-2N和Au/NU-3N保留了NU的形貌,粒径约为50 nm,表明即使经过后合成修饰,其骨架结构依然稳固。像差校正高角度环形暗场扫描透射电子显微镜(HAADF-STEM)图像(图2a)也显示了Au/NU、Au/NU-1N、Au/NU-2N和Au/NU-3N的清晰形貌,证实了它们保留了NU骨架的形貌。在Au/NU-2N的HAADF-STEM图像(图2b)中可以清晰地观察到孤立的亮点,证实了Au物种在整个框架中呈原子级分布,这可以最大限度地暴露催化活性位点。此外,框架的整体晶体结构仍然保持完整,如1.61 nm的晶面间距所示(图2b插图)。能量色散X射线(EDX)元素映射结果表明,Au物种在Au/NU-2N的框架中均匀分布(图2c)。此外,低氯含量证实了在Au/NU-2N的合成过程中氯的有效去除。综上所述,NU中引入的不同N-杂环配体能够实现Au物种的原子尺度调控,从而导致其分散性不同,并具有固有的Au配位基序。采用粉末X射线衍射(PXRD)技术对Au/NU、Au/NU-1N、Au/NU-2N和Au/NU-3N的晶体结构进行了表征。如图2d所示,NU的PXRD图谱与模拟结果吻合良好,证实了其成功合成。引入N-杂环配体并负载Au物种后,NU的特征PXRD衍射峰得以保留,表明其晶体结构在剧烈反应后仍保持稳定。Au/NU、Au/NU-1N和Au/NU-3N在38°和44°处出现了新的衍射峰,分别归属于金属Au的(111)和(200)晶面,证实了Au纳米颗粒的存在。
为了揭示Au/NU、Au/NU-1N、Au/NU-2N 和 Au/NU-3N中不同Au配位基元的结构,该团队进行了XPS分析。全谱结果显示均存在明显的N 1s和Au 4f信号,证明Au物种及含氮配体成功引入。高分辨XPS表明,Au/NU-2N中以Au+为主要价态,同时存在少量Au3+,说明AuCl4⁻未完全还原。Au 4f7/2结合能大小规律为Au/NU-2N > Au/NU-3N > Au/NU-1N(图2e),表明不同配体调控了Au的电子结构。其中,Au/NU-2N中原子分散的Au–N配位使其结合能最高,而Au/NU-1N和Au/NU-3N中以Au纳米颗粒为主,Au–Au相互作用占主导,从而降低结合能。O 1s光谱整体正移,说明Au–N配位及Au–O相互作用增强了电子抽取效应,降低了氧周围电子密度,且在Au/NU-2N 中最为显著。N 1s光谱进一步证实Au–N配位的存在,并表明2N在调控Au–N相互作用方面达到最佳平衡,从而有利于形成稳定的原子级Au–N/O配位结构。Au/NU-2N的N 1s XPS谱图可解卷积为五个不同的峰,分别位于399.3、400.0、400.6、401.7和401.1 eV,分别归属于C═N、Au–N、C–N、C–N–H和质子化的N+物种(图2f),表明存在Au–N配位基序。Au/NU-1N和Au/NU-3N的N 1s XPS谱图也显示出代表Au–N配位基序的峰。进一步分析表明,NU-1N、NU-2N 和 NU-3N 的主峰在锚定Au物种后发生正向偏移,进一步证实了其Au–N配位基序的存在。与Au/NU-2N相比,Au/NU-1N和Au/NU-3N的N 1s XPS峰分别出现负向和正向偏移(图2f)。这一结果源于引入的N-杂环配体导致其Au-N配位能力过弱或过强。因此,在Au/NU-2N中接枝2N可建立最佳的Au-N配位平衡,并促进原子级Au-N/O配位基序的分散,从而在Au物种和NU骨架之间形成有效的电子转移通道。
为进一步明确各样品中Au配位结构,该团队采用XANES和EXAFS进行分析。Au L3-edge XANES结果(图2g)显示,与Au箔相比,Au/NU、Au/NU-2N和Au/NU-3N均表现出增强的白线强度,表明存在氧化态Au。其中Au/NU-2N的白线强度最高,说明其Au物种具有更高氧化态,这与其原子分散的Au–N/O配位结构一致,并得到XPS结果的支持。相比之下,Au/NU-1N 的白线峰与Au箔几乎重合,表明其Au主要以金属态存在,呈现大尺寸纳米颗粒特征。FT-EXAFS 分析进一步揭示配位结构差异。Au/NU-2N在约1.68 Å处仅出现Au–N(O) 配位峰,且未检测到Au–Au信号,证明其Au以原子分散形式存在。而Au/NU、Au/NU-1N 和Au/NU-3N在2.4和3.0 Å处均出现明显Au–Au配位峰,说明存在Au纳米颗粒,同时伴随表面Au–O或Au–N/O配位结构。Au–N(O) 配位峰相较Au/NU出现负移,反映不同配体对Au–N配位环境的调控作用。EXAFS 拟合结果表明,Au/NU-2N中Au–N和Au–O的配位数分别约为1.9和2.8,说明每个Au原子由两个吡唑氮和三个水分子氧共同配位,形成稳定的原子级Au–N/O结构。而其他样品中同时存在Au–Au配位,表明其主要为纳米颗粒结构,其中 Au/NU-1N的Au–Au配位数较高,说明颗粒尺寸更大。波列变换分析进一步证实,Au/NU、Au/NU-1N 和 Au/NU-3N均在约10 Å⁻¹处出现Au–Au信号,而Au/NU-2N 仅在约5 Å⁻¹处出现Au–N(O) 信号,进一步证明其为原子分散结构。综上,引入含氮杂环配体可有效调控Au的配位结构,其中2N由于适中的配位能力,最有利于形成原子分散的Au–N/O配位结构,从而促进电子传输并最大化活性位点利用。
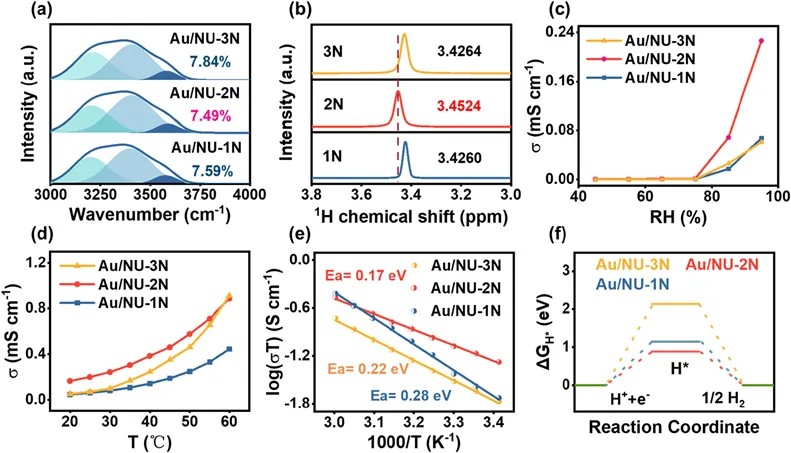
Fig. 3. (a) ATR-FTIR of Au/NU-1N, Au/NU-2N, and Au/NU-3N in water. (b) 1H NMR chemical shifts for 1N, 2N, and 3N in deionized water/DMSO-d6. (c) Humidity-dependent proton conductivity of Au/NU-1N, Au/NU-2N, and Au/NU-3N at 25 °C. (d) Temperature-dependent proton conductivities for Au/NU-1N, Au/NU-2N, and Au/NU-3N at 95% relative humidity. (e) Arrhenius plots of the proton conductivity of Au/NU-1N, Au/NU-2N, and Au/NU-3N. (f) Gibbs free energy diagrams of Au/NU-1N, Au/NU-2N, and Au/NU-3N toward photocatalytic hydrogen evolution.
引入的N-杂环配体的布朗斯台德碱性使其能够与水形成氢键。因此,通过Au配位基团,它们可以赋予Au/NU-1N、Au/NU-2N和Au/NU-3N不同的氢键网络,从而调控质子转移动力学,进而影响光催化析氢反应。为探究氢键网络结构,该团队采用ATR-FTIR分析(图3a)。O–H伸缩振动区(3000–4000 cm⁻¹)可分解为三类组分:约 3200 cm⁻¹ 的四配位氢键水(冰状水结构的特征峰)、约 3400 cm⁻¹ 的双配位氢键水(代表液态水)以及3600 cm⁻¹的自由水。定量拟合结果表明,Au/NU-2N 的自由水比例最低(7.49%),低于 Au/NU-1N(7.59%)和 Au/NU-3N(7.84%),说明其具有更强的氢键网络,使更多水分子参与氢键作用,有利于构建连续质子传输通道并降低传输能垒。1H NMR 表征(图3b)进一步验证了这一结论。与1N和 3N 相比,Au/NU-2N 中水质子信号发生明显下移,表明氢键增强导致质子去屏蔽效应增强。交流阻抗测试结果(图3c)表明,随着相对湿度由45%提升至95%,样品的阻抗显著降低,质子电导率提高。在95%湿度和25 °C条件下,Au/NU-2N的质子电导率达到2.26 × 10-4 S·cm⁻¹,明显高于Au/NU-1N(6.72 × 10-5 S·cm⁻¹)和Au/NU-3N(6.10 × 10-5 S·cm⁻¹),表明其氢键网络更有利于质子传输。温度升高同样促进质子迁移(图3d)。通过阿伦尼乌斯分析进一步确定了Au/NU-1N、Au/NU-2N和Au/NU-3N的质子传导活化能分别为0.28 eV、0.17 eV和0.22 eV(图3e)。所有值均低于0.4 eV的阈值,与格罗特胡斯机制相符。Au/NU-2N的活化能最低,这有利于质子通过氢键网络转移,并能为后续的光催化析氢反应提供充足的质子。这些结果证实,由N-杂环配体诱导的Au配位基序能够在Au/NU-1N、Au/NU-2N和Au/NU-3N中建立氢键网络,从而促进质子转移。值得注意的是,Au/NU-2N展现出最佳的氢键网络,为光催化析氢反应提供了充足的质子供应,使其位于催化位点附近。除质子供给外,催化剂对质子还原能力同样关键。计算得到H* 吸附自由能(ΔGH*)(图3f)分别为1.15、0.88 和2.13 eV,其中Au/NU-2N最接近理想值,表明其在热力学上更有利于析氢反应。综上,含氮配体诱导的Au配位结构能够构建氢键网络并调控质子传输,其中2N在氢键强度、质子导电性和还原动力学之间实现最佳平衡,从而为光催化析氢提供充足质子并显著提升反应效率。
Fig. 4. (a) UV–vis DRS, (b) EIS spectra, (c) transient photocurrent responses, and (d) steady-state PL spectra of NU, Au/NU, Au/NU-1N, Au/NU-2N, and Au/NU-3N. XPS spectra of (e) Au 4f and (f) N 1s of Au/NU-2N before and after light illumination. Time evolution of femtosecond TA spectra of (g) Au/NU-1N, (h) Au/NU-2N, and (i) Au/NU-3N in H2O upon 400 nm excitation, monitored over the 440–700 nm spectral window within an 8 ns display range. Evolution-Associated Difference Spectra (EADS) obtained from the global analysis based on a sequential model are shown in the lower panels below the corresponding TA spectra. The fitting time constants are shown in the legends.
NU中构建的Au配位结构显著影响材料的光电性能以及光生载流子的分离与传输行为。为此,该团队对样品进行了系统表征。UV–vis DRS结果(图4a)表明,与NU相比,Au/NU、Au/NU-1N和Au/NU-3N 在250–500 nm范围内表现出更强且更宽的吸收,并在532 nm处出现特征峰,归因于Au纳米颗粒的局域表面等离子体共振(LSPR)效应。相比之下,Au/NU-2N 未观察到LSPR特征峰,明确排除了Au纳米颗粒的存在,这与STEM、XPS和XAS结果一致。电化学测试进一步证实其优越的电荷动力学行为。EIS与瞬态光电流结果(图4b,c)表明,Au/NU-2N具有更低的电荷传输阻抗和更高的光电流响应,说明其电荷分离与传输效率显著提升。综上,Au/NU-2N 中的原子级 Au–N/O 配位结构不仅提供丰富的电子捕获位点以抑制载流子复合,还构建了高效的电荷传输通道,从而显著促进后续光催化反应。
为进一步探究Au配位结构对电荷分离与传输行为的影响,该团队进行了稳态光致发光(PL)与时间分辨瞬态光致发光(TR-PL)测试。如图4d所示,NU 及其配体修饰样品在480–510 nm处均表现出特征发射峰,而引入Au后(Au/NU、Au/NU-1N、Au/NU-2N、Au/NU-3N)均出现显著荧光淬灭。对于Au/NU、Au/NU-1N和Au/NU-3N,Au纳米颗粒的LSPR效应可增强局域电磁场,促进电子从骨架中抽取并抑制复合,这一点可由发射边红移(图4d插图)佐证。相比之下,Au/NU-2N中原子分散的Au–N/O配位结构构建了高效电子传输通道,使光生电子快速迁移至Au位点,从而更显著地淬灭荧光并提升电荷分离效率。TR-PL结果显示双指数衰减行为,其中τ₁对应快速电荷转移过程,τ₂反映激发态寿命。Au/NU-2N 的τ₂(5.15 ns)明显长于Au/NU、Au/NU-1N和Au/NU-3N,说明其载流子寿命得到有效延长,这归因于原子级Au–N/O配位结构促进电子传输并稳定载流子。同时,Au/NU-2N 具有最低的激发态占比(2.31%),结合其较高的光电流响应,表明其更有利于激子解离和后续电荷迁移。本质上,这源于原子分散的 Au–N–O 位点作为高效电子受体,能够快速捕获光生电子并抑制电子–空穴复合。
为进一步揭示电荷迁移机制,进行了原位XPS与EPR表征。如图4e所示,在光照条件下,Au/NU-1N、Au/NU-2N 和Au/NU-3N中Au 4f结合能均发生负移,说明Au位点可有效捕获光生电子。其中,Au/NU-2N 在90.6 eV处的Au 4f5/2峰面积显著降低,表明原子分散Au–N/O位点对电子的抽取能力更强,而其他样品因存在Au–Au配位(纳米颗粒)而该效应较弱。同时,N 1s结合能的负移(图4f)证明电子在含氮配体区域富集,说明N杂环配体充当电子桥梁,促进骨架向 Au 位点的电荷转移。EPR结果进一步提供直接证据。在g = 2.003处观察到光生电子信号,且随光照时间延长信号增强,表明电荷持续积累。其中,Au/NU-2N的信号增强最为显著,说明其电荷分离与传输效率最高。
飞秒瞬态吸收(fs-TA)光谱进一步揭示载流子动力学过程。NU在0.1 ps时表现为440–700 nm的基态漂白(GSB)信号,随后在皮秒尺度出现弱的激发态吸收(ESA),但在纳秒尺度迅速衰减,表明电荷易复合。Au/NU虽在初始阶段表现出增强的电荷分离信号,但后续仍存在明显复合,说明激子解离效率有限。相比之下,Au/NU-1N、Au/NU-2N 和 Au/NU-3N 在皮秒尺度均表现出显著的电荷分离特征(图4g–4i),其电荷分离时间常数约为2–5 ps,且电荷分离态寿命可达200–500 ps,随后形成稳定的自由载流子信号。其中,Au/NU-1N 和 Au/NU-2N具有更快的电荷转移(约2 ps)和激子解离过程(约290 ps),明显优于Au/NU-3N。综上,Au/NU-2N 中原子分散的Au–N/O配位结构不仅能够高效抽取骨架中的光生电子,还显著提升电荷分离与传输效率并延长载流子寿命,从而实现对光生电子的高效利用,最终促进光催化析氢反应。
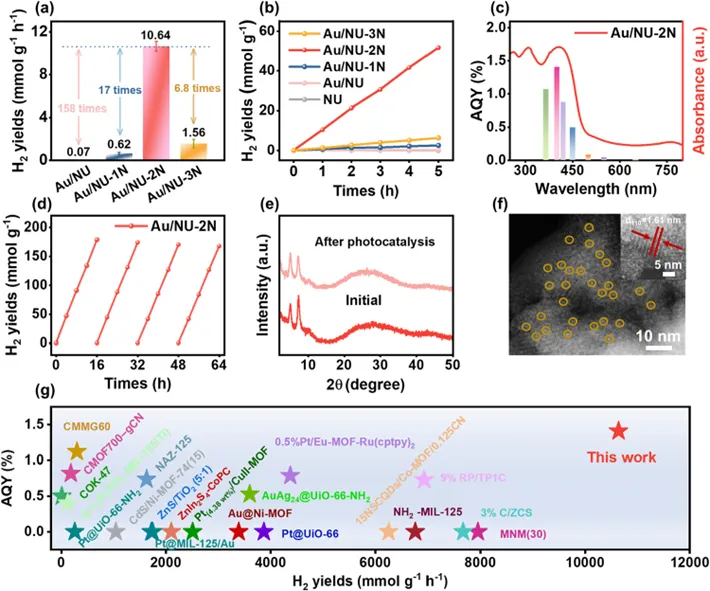
Fig. 5. (a) Photocatalytic hydrogen evolution rates of Au/NU, Au/NU-1N, Au/NU-2N, and Au/NU-3N, measured using ascorbic acid as a sacrificial agent. (b) Photocatalytic hydrogen evolution performance of NU, Au/NU, Au/NU-1N, Au/NU-2N, and Au/NU-3N upon 5 h irradiation. (c) The wavelength-dependent AQY of photocatalytic hydrogen evolution of Au/NU-2N. (d) Photocatalytic hydrogen evolution stability of Au/NU-2N upon 64 h irradiation. (e) PXRD patterns of Au/NU-2N before and after photocatalytic hydrogen evolution. (f) HAADF-STEM image of Au/NU-2N after 4 cycles of photocatalytic hydrogen evolution (orange circles represent Au single atoms). (g) Comparison of photocatalytic hydrogen evolution performance and AQY for Au/NU-2N and other MOF-based photocatalysts.
基于前述结构与电荷动力学分析,具有原子分散Au–N/O配位结构的Au/NU-2N同时具备优化的氢键网络和高效的电荷分离/传输能力,预示其优异的光催化析氢性能。为验证这一点,对所有样品在全光谱照射下进行了系统的光催化测试。如图5a、b所示,在优化Au负载量并以抗坏血酸为牺牲剂条件下,Au/NU-2N的析氢速率达到10.64 mmol gcat⁻¹ h⁻¹(或261.43 mmol gAu⁻¹ h⁻¹),分别较Au/NU、Au/NU-1N和Au/NU-3N提高158倍、17倍和6.8倍。波长依赖的表观量子效率(AQY)结果(图5c)与UV–vis 吸收一致,证明其具有宽光谱响应。在400 nm下AQY达1.41%,优于多数已报道MOF基光催化剂。稳定性测试表明,Au/NU-2N在连续运行64 h(4个循环)后几乎无性能衰减(图5d),而其他样品在数小时内即明显失活。结构表征进一步证明其优异稳定性。PXRD结果(图5e)显示反应前后结构基本一致,且未观察到Au聚集峰,说明Au–N/O配位结构得以保持。相比之下,其他样品虽保持骨架结构,但其Au纳米颗粒相关衍射峰减弱,表明活性位点发生局部无序化,这与其性能衰减相一致。STEM结果显示框架晶面间距(1.61 nm)保持不变(图5f 插图),且未观察到Au团簇或纳米颗粒,进一步证明Au仍以原子分散形式存在(图5f)。上述结果表明,Au/NU-2N中的Au–N/O原子级配位结构不仅显著提升催化活性,还赋予体系优异的稳定性,凸显了配体调控在构筑高效活性位点中的关键作用。值得指出的是,其析氢性能已优于多数已报道 MOF 基催化剂(图5g),展现出良好的应用潜力。