成都大学康辉、太原理工章日光,大连化物所刘岳峰Nature子刊丨反应诱导重构!Co-Mn界面位点驱动钴纳米簇“变身”精准调控CO₂加氢选择性
题目:Reaction-induced modification of Co nanoclusters driven by Co-Mn interfacial sites to control selectivity in CO2 hydrogenation
第一作者:Hui Kang,Rong Cao(共同一作)
通讯作者:Hui Kang(康辉,成都大学),Riguang Zhang(章日光,太原理工大学),Yuefeng Liu(刘岳峰,中国科学院大连化学物理研究所)
通讯单位:成都大学高等研究院,大连化物所洁净能源国家实验室,四川大学化学工程学院,太原理工大学化学化工学院,墨西拿大学ChiBioFarAm系
论文DOI:10.1038/s41467-026-70328-z
全文速览
在钴基纳米催化剂上精准调控反应选择性是催化科学的核心挑战之一。本研究报道了MnOₓ负载的2%钴纳米簇催化剂(2Co/MnOₓ)在CO₂加氢反应中发生的一场“反应诱导的表面重构”。在反应过程中,CO分子在Co-Mn界面解离生成的碳原子迁移并覆盖钴纳米簇表面,形成独特的碳修饰钴活性中心。这一原位重构戏剧性地逆转了产物选择性:CO/CH₄产物比从初始的0.89飙升至13.4,提升了15倍!通过系统的原位表征(DRIFTS、TPH、TPO、HRTEM)与DFT计算,研究揭示了这一重构的微观机制:MnOₓ的强亲氧性与Co的强亲碳性在界面处形成独特的Co-C-O-Mn桥式吸附构型,显著降低CO解离能垒;解离生成的碳原子高效迁移至Co表面,抑制了CO中间体的深度加氢。这一效应具有高度的特异性——仅存在于2Co/MnOₓ催化剂,在其他负载量或其他载体(TiO₂、SiO₂)上均未观察到。本研究展示了如何利用反应气氛诱导的结构演变实现对金属纳米催化剂表面性质的精准工程化,为抗CO中毒催化剂和CO₂高值转化催化剂的设计开辟了新路径。
背景介绍
钴纳米簇是费托合成、CO₂甲烷化、烷烃脱氢等众多工业催化过程的核心活性组分。这些反应通常具有结构敏感性。以费托合成为例,最佳钴粒径为6-8 nm,但出于金属利用率考虑,更小的纳米粒子更具吸引力。钴纳米簇具有卓越的H-H键和C-O键活化能力以及C-C偶联能力,是合成气制高附加值多碳化合物的广泛应用的活性中心。然而,甲烷化反应作为费托合成中的副反应,是需要被抑制的不期望过程。
反应过程中钴的碳化可调节催化选择性。钴纳米粒子的碳包覆和碳化钴(Co₂C)的形成是调控选择性的有效方法,特别是在费托合成中促进烯烃和含氧化合物的生成。然而,这些转变同时抑制水煤气变换反应,且与合成气相比,在CO₂/H₂进料下表现出不稳定性,使其不适用于直接的CO₂制烯烃生产。在CO₂加氢过程中提高CO/CH₄比为后续以CO为关键平台分子的费托合成和甲醇工业提供了重要基础,这取决于催化剂表面微观结构的可控调制。
反应驱动的结构变化为调控催化剂结构、克服这些限制提供了一种具有工业潜力的替代方法。揭示驱动这些气体诱导结构变化的复杂机制是催化剂设计的一种开创性方法,为开发定制催化剂提供了新视角。对一个反应失活的催化剂结构,可能是另一个反应的优异活性位点。换言之,战略性地设计反应气氛以诱导目标反应所需的具体结构,是一种具有工业潜力的宝贵催化剂设计原则。
通过调整催化剂组成(载体类型和Co负载量)和不同气体(如纯CO₂、CO和H₂,以及CH₄、CO或CO₂与H₂混合)下的预处理条件,我们揭示了控制Co基催化剂在反应过程中结构演变及CO₂加氢(逆水煤气变换反应)中选择性调控的开关。值得注意的是,2Co/MnOₓ催化剂在CO₂加氢过程中表现出显著、快速的表面碳物种形成(而非碳化物CoₓC生成),使主要产物从CH₄转向CO。全面的表面结构表征和原位实验揭示,碳重构的Co表面决定性地削弱了CO吸附和加氢能力。这种结构演变关键依赖于MnOₓ载体的强亲氧性和Co金属的亲碳性,这两者共同促进了Co/MnO界面处Co-C-O-Mn构型的CO化学吸附。吸附的CO随后发生歧化反应生成C原子,这些C原子迁移覆盖Co纳米簇表面。
本研究超越了现有的碳改性钴纳米催化剂(包括Mn促进的材料),代表了诱导钴纳米簇可控气体诱导改性的新进展。本研究采用可控的气体诱导策略来改性活性金属的表面性质,阐明演变结构如何从机理上影响反应性能,并进一步强调此类气体诱导策略在纳米甚至原子水平上调控催化剂表面结构的关键意义。
本文亮点
- 发现反应诱导的表面重构新机制:首次报道在温和CO₂加氢反应条件下(400°C),2Co/MnOₓ催化剂发生碳诱导的钴表面重构,而非传统的碳化物形成或积碳失活。这一重构戏剧性地逆转了反应选择性,CO/CH₄比从0.89飙升至13.4,提升了15倍!该效应具有高度特异性,仅存在于特定Co负载量(2%)和MnOₓ载体组合。
- 揭示Co-Mn界面协同的原子级机制:结合原位光谱(CO-DRIFTS、NO-DRIFTS)和DFT计算,首次实验证实Co/MnO界面形成独特的Co-C-O-Mn μ²-桥式羰基构型。该构型充分利用了Co的亲碳性和Mn的亲氧性,将CO解离能垒从2.63 eV(纯Co)降至2.05 eV,同时解离生成的O在MnO表面以近乎无垒(0.06 eV)的方式被CO或H清除,形成高效的CO→C原子转化流水线。
- 建立“碳修饰钴”活性中心新概念:与文献报道的Co₂C碳化物或碳包覆Co纳米粒子不同,本工作发现的表面聚合碳物种修饰的Co纳米簇具有独特性质:它抑制了CO的过度加氢(甲烷化),但保留了CO₂转化活性,且表现出优异的稳定性。TPH、TPO和循环反应-再生实验确证,聚合碳物种是可逆生成和消除的“选择性开关”,通过调控反应气氛即可实现选择性的可逆切换。
图文解析
图1:反应诱导的Co/MnOₓ催化剂选择性演变
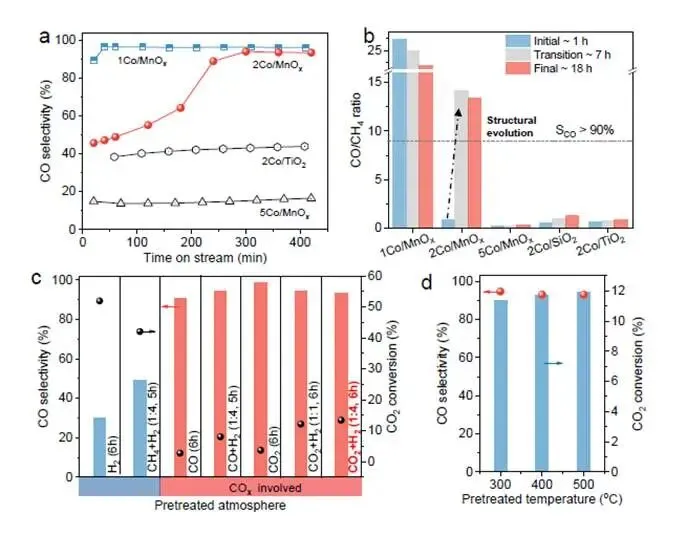
要点:研究者系统评估了不同Co负载量和不同载体的催化剂在CO₂加氢反应中的性能演变。
- 2Co/MnOₓ展现戏剧性选择性翻转:在400°C反应过程中,2Co/MnOₓ催化剂的CO选择性在5小时内从45.7%飙升至94.0%(图1a),CO/CH₄比从初始的0.89增至18小时后的13.4(图1b)。这是前所未有的选择性逆转现象!
- 效应具有高度特异性:1Co/MnOₓ虽有提升但幅度较小,5Co/MnOₓ、2Co/TiO₂和2Co/SiO₂均未观察到明显选择性变化(图1a-b)。表明2% Co负载量与MnOₓ载体的组合是实现该效应的关键。
- COₓ气氛是重构的“触发器”:通过七种不同气体预处理(图1c),发现只有含CO或CO₂的气氛能诱导高CO选择性(>90%),而H₂或CH₄+H₂处理无效。这明确指向CO是驱动结构演变的活性物种。
- 重构是温度独立的终态:将催化剂在不同温度(300-500°C)下反应18小时后,再在400°C测试,性能几乎相同(图1d)。表明最终重构状态由气氛决定,温度仅影响重构速率。
解析:图1确立了2Co/MnOₓ催化剂的独特性:它在CO₂加氢反应中发生自发的结构演变,产物选择性从甲烷转向CO。COₓ气氛是这一演变的必要条件,而特定Co负载量和MnOₓ载体则是实现高效重构的充分条件。这一发现为后续机理研究指明了方向:谁在变?怎么变?为什么是2Co/MnOₓ?
图2:重构的本质——碳诱导的Co表面改性
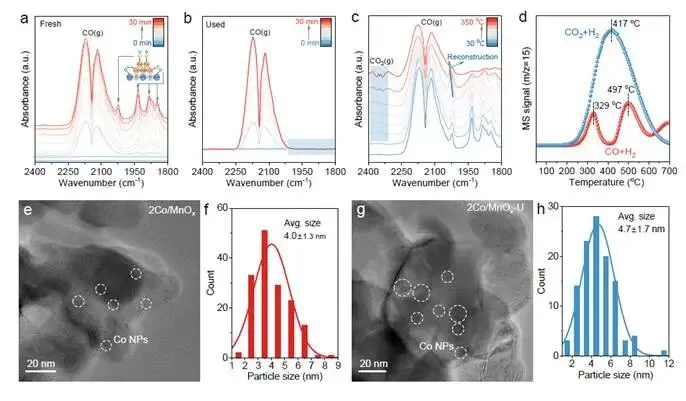
要点:研究者运用多种表征技术,深入揭示了催化剂在反应前后的结构变化。
- CO吸附能力彻底丧失:新鲜2Co/MnOₓ催化剂表现出丰富的CO吸附位点,包括线性吸附(~2022 cm⁻¹)和桥式吸附(<2000 cm⁻¹)(图2a)。然而,反应后催化剂(2Co/MnOₓ-U)表面完全检测不到CO吸附信号(图2b),表明Co活性中心发生了深刻的结构改变。
- 升温诱导CO吸附蓝移:升温CO-DRIFTS显示,随着温度升高,线性CO吸附峰发生蓝移(图2c),这违背了常规的覆盖度依赖红移规律³²⁻³³,暗示钴中心在CO处理过程中发生了内在结构变化,逐步削弱CO吸附。
- 排除粒径和碳化物机制:TEM统计显示,反应后Co粒径仅从4.0 nm微增至4.7 nm(图2e-h),不足以解释选择性巨变。XPS和NAP-XPS均未检测到CoCₓ物种(283.1 eV)(图S11),排除了碳化物机制。
- CO是关键活性物种:CO+H₂-TPR在417°C出现活性陡降,而CO₂+H₂-TPR在更高温出现新峰(图2d),表明重构后的钴中心提高了CO加氢能垒,CO可能是驱动重构的关键物种。
解析:图2通过排除法(排除粒径变化、排除碳化物生成)和正向证据(CO吸附行为改变、CO-TPR活性变化),将重构的本质指向碳物种对Co表面的修饰。CO在反应过程中与Co表面发生强相互作用,改变了其电子性质和吸附能力,从而抑制了甲烷化路径。
图3:表面碳物种的确证与可逆调控
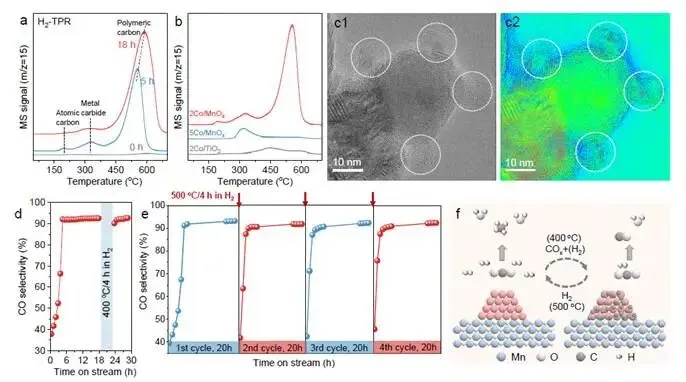
要点:研究者通过程序升温表面反应和TEM,直接捕获了表面碳物种的证据,并实现了选择性的可逆调控。
- TPR/TPO确证表面碳沉积:反应后2Co/MnOₓ的TPR谱显示三个CH₄生成峰(图3a):197°C(高活性表面原子碳)、325°C(金属-碳物种)、550-590°C(聚合碳物种)⁴⁰⁻⁴¹。TPO结果与之对应(图S12)。随着反应时间延长,各碳物种含量均增加。
- 碳沉积的载体和负载量依赖性:5Co/MnOₓ和2Co/TiO₂反应后表面碳物种丰度显著低于2Co/MnOₓ(图3b),与它们微弱的选择性变化一致。
- HRTEM直接观测到层状石墨碳:反应后2Co/MnOₓ表面清晰可见层状石墨碳物种(图3c1-c2,S18-19),确证碳沉积的发生。但活性测试表明催化剂并未失活,说明碳层具有多孔性,允许反应物接触Co表面,同时改变了表面反应性。
- 选择性可逆切换的“开关”:反应18 h后的2Co/MnOₓ在400°C H₂处理4 h后,高CO选择性保持不变(图3d);但500°C H₂处理4 h后,催化剂完全恢复至初始的高CH₄选择性(图3e)。TPH表明500°C H₂可彻底清除聚合碳物种。循环反应-还原-反应实验(图3f)确证:聚合碳物种是可逆生成和消除的“选择性开关”。
解析:图3是全文的“破案时刻”。TPR/TPO和HRTEM直接证明了反应过程中聚合碳物种在Co表面沉积。更关键的是,通过控制H₂处理温度,可以实现选择性的可逆切换,形成完美的闭环证据链:聚合碳沉积 → 高CO选择性;聚合碳清除 → 恢复高CH₄选择性。这确凿无疑地证明:表面聚合碳物种是驱动选择性翻转的根本原因。
图4:Co/MnO界面的关键作用——亲氧与亲碳的完美协同
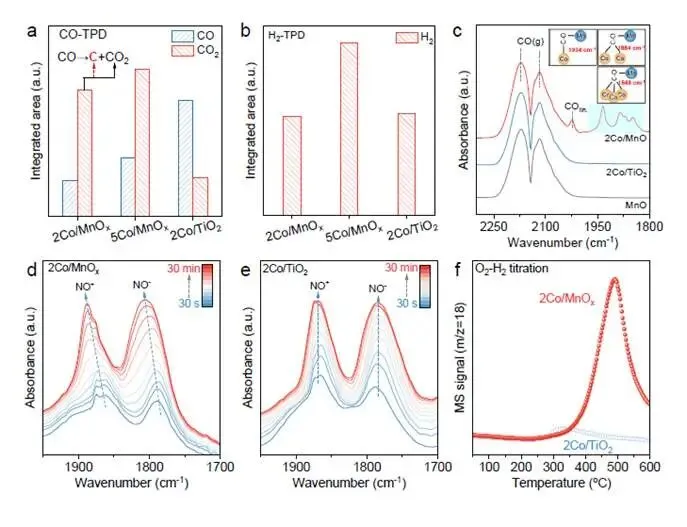
要点:为什么是MnOₓ?为什么是2% Co?图4揭示了Co/MnO界面的独特性质。
- MnOₓ促进CO歧化:CO-TPD定量分析显示,2Co/MnOₓ和5Co/MnOₓ的CO₂脱附量远高于2Co/TiO₂(图4a),表明MnOₓ载体显著增强了CO歧化反应(2CO → CO₂ + C)能力,这是表面碳的来源。
- Co负载量调控碳去除:H₂-TPD显示5Co/MnOₓ的H₂脱附量远高于2Co/MnOₓ(图4b),意味着更高Co负载量提供更多H₂解离位点,加速碳物种的氢化去除,因此净碳沉积速率反而更低。这完美解释了为什么2%是“黄金负载量”——碳生成与碳去除达到最优平衡。
- Co/MnO界面形成独特吸附构型:CO-DRIFTS中,2Co/MnOₓ在1884和1848 cm⁻¹出现独特吸收峰(图4c),归属为Co-C-O-Mn μ²-桥式羰基⁵²⁻⁵⁴。该构型在MnOₓ和2Co/TiO₂上均不存在,是Co/MnO界面的“指纹信号”。
- MnOₓ的亲氧性实验证实:NO-DRIFTS中,随着NO覆盖度增加,2Co/MnOₓ的NO吸附峰发生明显蓝移(图4d),而2Co/TiO₂无此现象(图4e)。蓝移源于MnOₓ的亲氧性导致Co表面部分氧化,削弱了NO吸附。O₂-H₂滴定进一步证实2Co/MnOₓ具有更多氧吸附位点(图4f)。
解析:图4揭示了2Co/MnOₓ催化剂的“超能力”来源:Co/MnO界面是集“亲碳”与“亲氧”于一体的完美反应器。Co提供碳亲和位点吸附CO,Mn提供氧亲和位点稳定解离的O*,二者通过Co-C-O-Mn桥式构型协同作用,将CO高效转化为C原子和CO₂(或H₂O)。同时,适当的Co负载量(2%)平衡了碳生成(界面歧化)与碳去除(Co表面H₂解离),实现持续可控的表面碳修饰。
图5:DFT计算揭示原子级机理
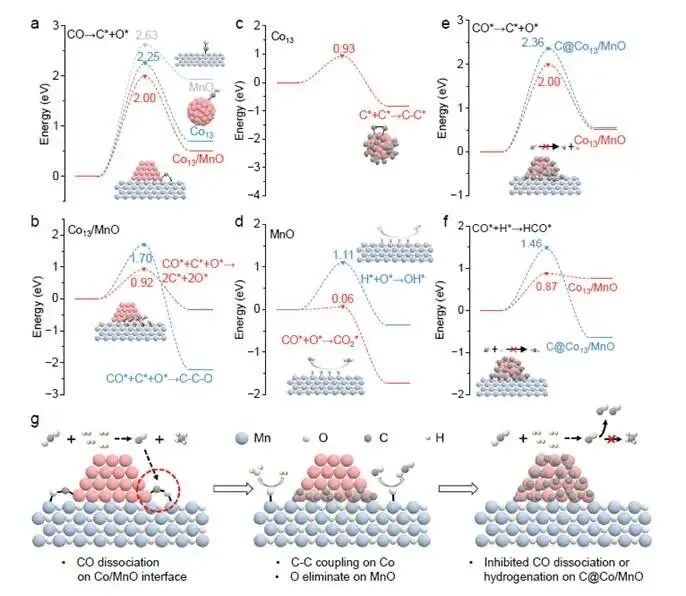
要点:DFT计算为实验观测提供了原子尺度的理论支撑,完美阐释了整个反应路径。
- 界面协同降低CO解离能垒:CO解离能垒计算(图5a)显示,在Co₁₃/MnO界面上,C-O键断裂能垒为2.05 eV,显著低于纯Co₁₃簇(2.63 eV)和纯MnO(2.25 eV)。这表明Co-Mn协同作用是CO高效解离的关键。
- 界面生成C原子高效迁移:在Co₁₃/MnO界面,第二个CO分子的解离能垒仅0.92 eV,远低于C-C偶联能垒(1.70 eV)(图5b)。而纯Co₁₃表面C-C偶联能垒为0.93 eV(图5c)。这意味着在界面上,CO解离生成C原子是主导路径,且C原子倾向于迁移而非偶联。DFT还显示C原子从界面迁移至Co表面的能垒仅0.82 eV,在Co表面迁移能垒仅0.21 eV(图S33-34),迁移极其容易。
- MnO表面O*高效清除:CO* + O* → CO₂的能垒仅0.06 eV(图5d),几乎是自发反应;H + O* → OH的能垒也仅1.11 eV。这确保了界面活性位点不被O毒化,持续运转。
- 碳修饰抑制CO活化和加氢:在碳修饰的C@Co₁₃/MnO界面上,CO解离能垒升至2.36 eV,CO加氢能垒升至1.46 eV(图5e-f),均高于未修饰界面(2.00 eV和0.87 eV)。这直接证明了表面碳物种抑制了CO的进一步活化和加氢,从而抑制甲烷化,促进CO生成。
- 完整机理图景:图5g总结了整个反应诱导重构的机制:CO在Co/MnO界面通过Co-C-O-Mn构型高效解离 → C原子迁移覆盖Co表面 → O被CO或H快速清除 → 碳修饰的Co表面抑制CO加氢 → 产物选择性从CH₄转向CO。
解析:图5的DFT计算与实验观测完美吻合,构建了从分子尺度到宏观催化性能的完整桥梁。计算不仅解释了“为什么2Co/MnOₓ会发生重构”(界面协同降低解离能垒),还解释了“重构后为什么选择性改变”(碳修饰抑制加氢),并阐释了“为什么2%是最优负载量”(碳生成与碳去除的动力学平衡)。
总结与展望
本研究系统揭示了Co/MnOₓ催化剂在CO₂加氢过程中反应诱导的结构演变与选择性调控机制。核心发现与结论如下:
- 反应诱导的碳修饰重构:2Co/MnOₓ催化剂在CO₂加氢反应中发生独特的表面重构,CO分子在Co-Mn界面解离生成的碳原子迁移并覆盖Co纳米簇表面,形成聚合碳物种修饰的Co活性中心。这一重构使CO/CH₄产物比从0.89提升至13.4,提升了15倍。
- Co-Mn界面的原子级协同机制:MnOₓ的强亲氧性与Co的强亲碳性在界面处形成独特的Co-C-O-Mn桥式吸附构型,将CO解离能垒从2.63 eV降至2.05 eV。解离生成的O在MnO表面以近乎无垒的方式被CO或H清除,形成高效的CO→C原子转化流水线。
- 碳生成与碳去除的动态平衡决定重构速率:Co负载量调控表面H浓度,进而控制碳去除能力。2% Co实现碳生成(界面歧化)与碳去除(H₂解离)的最优平衡,是该效应的“黄金负载量”。
- 可逆调控的选择性“开关”:表面聚合碳物种是可逆生成和清除的。通过调控反应气氛(COₓ诱导重构,500°C H₂再生),可实现CO₂加氢选择性的可逆切换,为催化剂原位再生和智能调控提供了新思路。
本研究提出的反应诱导表面重构策略,超越了传统的碳化物或碳包覆改性范式,为设计抗CO中毒催化剂和CO₂高值转化催化剂开辟了新路径。未来可将该策略拓展至其他金属-载体组合,探索不同反应气氛诱导的结构演变,实现催化剂表面性质的精准工程化。
通讯作者简介
Hui Kang(康辉),成都大学高等研究院特聘研究员,硕士生导师。2022博士毕业于四川大学,师从储伟教授;2024年加入成都大学。主要从事CO/CO₂催化转化、原位表征、催化剂动态结构演变研究。以第一/通讯作者在Nature Catal., Nature Commun., Chem. Soc. Rev., Appl. Catal. B等期刊发表论文多篇。主持国家自然科学基金青年项目、四川省自然科学基金等。电子邮箱:kanghui@cdu.edu.cn。
Riguang Zhang(章日光),博士,三级教授,博士生导师,国家重点实验室副主任。
2009~2013年于太原理工大学获得博士学位;2006~至今年毕业留校在太原理工大学煤化工研究所工作。2016/12–2017/12,美国怀俄明大学石油与化工系国家公派访问学者。2006年10月留校工作任助教、讲师,2013年7月特殊破格聘任教授。2018年8月任职煤科学与技术教育部和山西省重点实验室副主任,2021年1月任职省部共建煤基能源清洁高效利用国家重点实验室副主任。
2014年入选山西省第二批青年拔尖人才,山西省高等学校优秀青年学术带头人,2014年获山西省优秀博士论文,2014年山西省高校“131”领军人才,2016年获山西省学术技术带头人,2018年山西省三晋英才拔尖骨干人才,2021年山西省杰青项目获得者。任职期间作为项目负责人承担国家自然科学基金区域创新发展联合基金重点项目1项、重点新材料研发及应用国家科技重大专项(科技创新2030重大项目)任务1项、国家自然科学基金面上项目4项、国家重点研发计划项目子课题1项、国家自然科学基金青年基金1项,山西省自然科学基金杰青项目1项;同时,主持省级和学校等各类项目10余项。以第一完成人获2024年中国发明协会发明创业奖创新奖二等奖和2023年山西省教学成果奖(高等教育研究生)二等奖,第二完成人2020年山西省科学技术奖自然科学一等奖;截止2025年2月,在国内外权威学术期刊Nat. Common. (3篇)、Angew. Chem. Int. Ed. (2篇)、PNAS (1篇)、ACS Catal. (6篇)、Chem. Eng. Sci. (15篇)、J. Catal. (4篇)、Appl. Catal. B: Environ. (6篇)、Chem. Eng. J. (12篇)等发表SCI收录学术论文260篇,SCI正面他引近4000余次。授权国家发明专利7项。任职《煤炭转化》副主编、《燃料化学学报》、《Green Energy & Environment》青年编委,山西省化学会副秘书长。电子邮箱:zhangriguang@tyut.edu.cn。
Yuefeng Liu(刘岳峰),博士毕业于法国斯特拉斯堡大学,致力于碳资源小分子清洁高值化利用中的应用基础研究;开发的碳基精脱硫剂在宝武集团宝山基地、中南钢铁等企业焦炉煤气系统精脱硫改造工程中工业应用。先后主持国家自然科学基金(包括重大研究计划培育项目、面上基金)、辽宁省自然科学基金以及大连市高层次人才创新支持计划等省部级、企业横向项目等多项;作为子课题负责人参与国家重点研发计划项目2项。发表SCI期刊论文120余篇,SCI引用超过9000次,H因子55 (Google scholar);其中第一作者/通讯作者论文90余篇,包括国际著名学术期刊Nature Catalysis, Angew Chem Int Ed, J Am Chem Soc (2篇), Nature Commun (4篇), Science Adv, Adv Mater, Chem Rev, Chem Soc Rev,ACS Catal (10篇), Appl Catal B: Environ (5篇)等。申请国际PCT专利2件,并获授权发明专利10件(美国2件,欧洲2件,中国6件)。连续入选全球前2%顶尖科学家榜单(2022-2024年度);担任中国颗粒学会青年理事,Journal of Energy Chemistry期刊(中国科学院分区1区)的客座编辑/青年编委,Chemical Synthesis、Carbon Future 和《低碳化学与化工》期刊青年编委。获中国颗粒学会“青年颗粒学奖”(2022)和大连市“青年科技之星”称号(2017);入选辽宁省“兴辽英才”青年拔尖人才计划(2019)和中国科学院青年促进会会员(2018)等。电子邮箱:yuefeng.liu@dicp.ac.cn。
声明:文章内容仅代表本人观点,本文数据来源于Nature Communications论文(DOI: 10.1038/s41467-026-70328-z),图片版权归原作者所有。如有侵权,请联系后台,感谢支持,欢迎批评指正!
欢迎各位老师同学们在公众号上投稿分享课题组研究成果,不限期刊和发表时间!后台联系即可。
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
